Jump to
S1C2
Energy calculated at B97D3/6-31G*
| | hartrees |
|---|
| Energy at 0K | -193.328352 |
| Energy at 298.15K | -193.332176 |
| HF Energy | -193.328352 |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B97D3/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3208 |
3145 |
0.02 |
|
|
|
| 2 |
A1 |
3183 |
3121 |
32.30 |
|
|
|
| 3 |
A1 |
3162 |
3100 |
11.27 |
|
|
|
| 4 |
A1 |
1476 |
1447 |
14.57 |
|
|
|
| 5 |
A1 |
1375 |
1348 |
21.65 |
|
|
|
| 6 |
A1 |
1133 |
1111 |
0.03 |
|
|
|
| 7 |
A1 |
1054 |
1033 |
0.13 |
|
|
|
| 8 |
A1 |
936 |
918 |
0.39 |
|
|
|
| 9 |
A1 |
833 |
817 |
0.30 |
|
|
|
| 10 |
A2 |
852 |
835 |
0.00 |
|
|
|
| 11 |
A2 |
684 |
671 |
0.00 |
|
|
|
| 12 |
A2 |
498 |
488 |
0.00 |
|
|
|
| 13 |
B1 |
844 |
827 |
4.85 |
|
|
|
| 14 |
B1 |
787 |
772 |
0.92 |
|
|
|
| 15 |
B1 |
663 |
650 |
75.25 |
|
|
|
| 16 |
B1 |
486 |
476 |
12.31 |
|
|
|
| 17 |
B2 |
3200 |
3137 |
22.82 |
|
|
|
| 18 |
B2 |
3171 |
3109 |
2.44 |
|
|
|
| 19 |
B2 |
1522 |
1492 |
0.47 |
|
|
|
| 20 |
B2 |
1277 |
1252 |
0.03 |
|
|
|
| 21 |
B2 |
1205 |
1181 |
0.71 |
|
|
|
| 22 |
B2 |
1063 |
1042 |
13.00 |
|
|
|
| 23 |
B2 |
924 |
906 |
5.33 |
|
|
|
| 24 |
B2 |
32 |
31 |
4.94 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16782.5 cm
-1
Scaled (by 0.9804) Zero Point Vibrational Energy (zpe) 16453.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B97D3/6-31G*
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
1.192 |
| C2 |
0.000 |
1.175 |
0.356 |
| C3 |
0.000 |
0.743 |
-0.952 |
| C4 |
0.000 |
-0.743 |
-0.952 |
| C5 |
0.000 |
-1.175 |
0.356 |
| H6 |
0.000 |
0.000 |
2.281 |
| H7 |
0.000 |
2.203 |
0.709 |
| H8 |
0.000 |
1.363 |
-1.847 |
| H9 |
0.000 |
-1.363 |
-1.847 |
| H10 |
0.000 |
-2.203 |
0.709 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.4423 | 2.2693 | 2.2693 | 1.4423 | 1.0896 | 2.2553 | 3.3302 | 3.3302 | 2.2553 |
C2 | 1.4423 | | 1.3781 | 2.3222 | 2.3509 | 2.2558 | 1.0863 | 2.2108 | 3.3609 | 3.3968 | C3 | 2.2693 | 1.3781 | | 1.4860 | 2.3222 | 3.3181 | 2.2115 | 1.0882 | 2.2879 | 3.3820 | C4 | 2.2693 | 2.3222 | 1.4860 | | 1.3781 | 3.3181 | 3.3820 | 2.2879 | 1.0882 | 2.2115 | C5 | 1.4423 | 2.3509 | 2.3222 | 1.3781 | | 2.2558 | 3.3968 | 3.3609 | 2.2108 | 1.0863 | H6 | 1.0896 | 2.2558 | 3.3181 | 3.3181 | 2.2558 | | 2.7068 | 4.3473 | 4.3473 | 2.7068 | H7 | 2.2553 | 1.0863 | 2.2115 | 3.3820 | 3.3968 | 2.7068 | | 2.6900 | 4.3870 | 4.4059 | H8 | 3.3302 | 2.2108 | 1.0882 | 2.2879 | 3.3609 | 4.3473 | 2.6900 | | 2.7258 | 4.3870 | H9 | 3.3302 | 3.3609 | 2.2879 | 1.0882 | 2.2108 | 4.3473 | 4.3870 | 2.7258 | | 2.6900 | H10 | 2.2553 | 3.3968 | 3.3820 | 2.2115 | 1.0863 | 2.7068 | 4.4059 | 4.3870 | 2.6900 | |
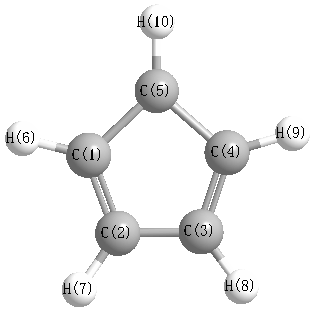 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
107.322 |
|
C1 |
C2 |
H7 |
125.432 |
| C1 |
C5 |
C4 |
107.322 |
|
C1 |
C5 |
H10 |
125.432 |
| C2 |
C1 |
C5 |
108.687 |
|
C2 |
C1 |
H6 |
125.657 |
| C2 |
C3 |
C4 |
108.334 |
|
C2 |
C3 |
H8 |
127.055 |
| C3 |
C2 |
H7 |
127.246 |
|
C3 |
C4 |
C5 |
108.334 |
| C3 |
C4 |
H9 |
124.611 |
|
C4 |
C3 |
H8 |
124.611 |
| C4 |
C5 |
H10 |
127.246 |
|
C5 |
C1 |
H6 |
125.657 |
| C5 |
C4 |
H9 |
127.055 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.123 |
|
|
|
| 2 |
C |
-0.149 |
|
|
|
| 3 |
C |
-0.135 |
|
|
|
| 4 |
C |
-0.135 |
|
|
|
| 5 |
C |
-0.149 |
|
|
|
| 6 |
H |
0.145 |
|
|
|
| 7 |
H |
0.134 |
|
|
|
| 8 |
H |
0.140 |
|
|
|
| 9 |
H |
0.140 |
|
|
|
| 10 |
H |
0.134 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.053 |
0.053 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.218 |
0.000 |
0.000 |
| y |
0.000 |
-27.093 |
0.000 |
| z |
0.000 |
0.000 |
-25.304 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-6.020 |
0.000 |
0.000 |
| y |
0.000 |
1.668 |
0.000 |
| z |
0.000 |
0.000 |
4.352 |
|
| Polar |
|---|
| 3z2-r2 | 8.704 |
|---|
| x2-y2 | -5.125 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.747 |
0.000 |
0.000 |
| y |
0.000 |
8.114 |
0.000 |
| z |
0.000 |
0.000 |
8.875 |
<r2> (average value of r
2) Å
2
| <r2> |
88.201 |
| (<r2>)1/2 |
9.392 |
Jump to
S1C1
Energy calculated at B97D3/6-31G*
| | hartrees |
|---|
| Energy at 0K | -193.328350 |
| Energy at 298.15K | |
| HF Energy | -193.328350 |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B97D3/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3207 |
3144 |
0.05 |
|
|
|
| 2 |
A1 |
3199 |
3136 |
22.85 |
|
|
|
| 3 |
A1 |
3165 |
3103 |
2.22 |
|
|
|
| 4 |
A1 |
1552 |
1521 |
0.00 |
|
|
|
| 5 |
A1 |
1435 |
1407 |
2.47 |
|
|
|
| 6 |
A1 |
1133 |
1111 |
0.00 |
|
|
|
| 7 |
A1 |
1073 |
1052 |
7.16 |
|
|
|
| 8 |
A1 |
1030 |
1010 |
10.39 |
|
|
|
| 9 |
A1 |
834 |
818 |
0.28 |
|
|
|
| 10 |
A2 |
856 |
839 |
0.00 |
|
|
|
| 11 |
A2 |
789 |
774 |
0.00 |
|
|
|
| 12 |
A2 |
512 |
502 |
0.00 |
|
|
|
| 13 |
B1 |
838 |
822 |
5.67 |
|
|
|
| 14 |
B1 |
687 |
673 |
8.81 |
|
|
|
| 15 |
B1 |
657 |
644 |
68.24 |
|
|
|
| 16 |
B1 |
475 |
465 |
10.64 |
|
|
|
| 17 |
B2 |
3183 |
3121 |
29.17 |
|
|
|
| 18 |
B2 |
3167 |
3105 |
14.66 |
|
|
|
| 19 |
B2 |
1386 |
1359 |
34.15 |
|
|
|
| 20 |
B2 |
1277 |
1251 |
0.00 |
|
|
|
| 21 |
B2 |
1203 |
1179 |
2.17 |
|
|
|
| 22 |
B2 |
963 |
945 |
0.56 |
|
|
|
| 23 |
B2 |
913 |
895 |
2.51 |
|
|
|
| 24 |
B2 |
92i |
90i |
7.38 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16719.8 cm
-1
Scaled (by 0.9804) Zero Point Vibrational Energy (zpe) 16392.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B97D3/6-31G*
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
1.231 |
| C2 |
0.000 |
1.130 |
0.394 |
| C3 |
0.000 |
0.684 |
-1.010 |
| C4 |
0.000 |
-0.684 |
-1.010 |
| C5 |
0.000 |
-1.130 |
0.394 |
| H6 |
0.000 |
0.000 |
2.317 |
| H7 |
0.000 |
2.170 |
0.718 |
| H8 |
0.000 |
1.343 |
-1.875 |
| H9 |
0.000 |
-1.343 |
-1.875 |
| H10 |
0.000 |
-2.170 |
0.718 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.4059 | 2.3435 | 2.3435 | 1.4059 | 1.0861 | 2.2298 | 3.3840 | 3.3840 | 2.2298 |
C2 | 1.4059 | | 1.4734 | 2.2941 | 2.2593 | 2.2302 | 1.0893 | 2.2791 | 3.3562 | 3.3153 | C3 | 2.3435 | 1.4734 | | 1.3687 | 2.2941 | 3.3971 | 2.2786 | 1.0871 | 2.2043 | 3.3364 | C4 | 2.3435 | 2.2941 | 1.3687 | | 1.4734 | 3.3971 | 3.3364 | 2.2043 | 1.0871 | 2.2786 | C5 | 1.4059 | 2.2593 | 2.2941 | 1.4734 | | 2.2302 | 3.3153 | 3.3562 | 2.2791 | 1.0893 | H6 | 1.0861 | 2.2302 | 3.3971 | 3.3971 | 2.2302 | | 2.6957 | 4.4020 | 4.4020 | 2.6957 | H7 | 2.2298 | 1.0893 | 2.2786 | 3.3364 | 3.3153 | 2.6957 | | 2.7210 | 4.3661 | 4.3397 | H8 | 3.3840 | 2.2791 | 1.0871 | 2.2043 | 3.3562 | 4.4020 | 2.7210 | | 2.6866 | 4.3661 | H9 | 3.3840 | 3.3562 | 2.2043 | 1.0871 | 2.2791 | 4.4020 | 4.3661 | 2.6866 | | 2.7210 | H10 | 2.2298 | 3.3153 | 3.3364 | 2.2786 | 1.0893 | 2.6957 | 4.3397 | 4.3661 | 2.7210 | |
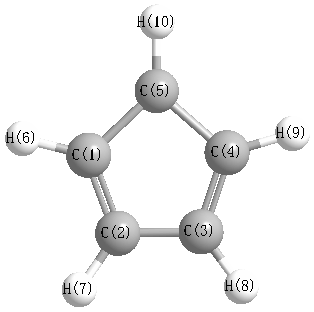 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
108.967 |
|
C1 |
C2 |
H7 |
126.207 |
| C1 |
C5 |
C4 |
108.967 |
|
C1 |
C5 |
H10 |
126.207 |
| C2 |
C1 |
C5 |
106.944 |
|
C2 |
C1 |
H6 |
126.528 |
| C2 |
C3 |
C4 |
107.561 |
|
C2 |
C3 |
H8 |
125.184 |
| C3 |
C2 |
H7 |
124.826 |
|
C3 |
C4 |
C5 |
107.561 |
| C3 |
C4 |
H9 |
127.255 |
|
C4 |
C3 |
H8 |
127.255 |
| C4 |
C5 |
H10 |
124.826 |
|
C5 |
C1 |
H6 |
126.528 |
| C5 |
C4 |
H9 |
125.184 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.152 |
|
|
|
| 2 |
C |
-0.127 |
|
|
|
| 3 |
C |
-0.144 |
|
|
|
| 4 |
C |
-0.144 |
|
|
|
| 5 |
C |
-0.127 |
|
|
|
| 6 |
H |
0.133 |
|
|
|
| 7 |
H |
0.144 |
|
|
|
| 8 |
H |
0.136 |
|
|
|
| 9 |
H |
0.136 |
|
|
|
| 10 |
H |
0.144 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.045 |
0.045 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.220 |
0.000 |
0.000 |
| y |
0.000 |
-25.310 |
0.000 |
| z |
0.000 |
0.000 |
-27.086 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-6.022 |
0.000 |
0.000 |
| y |
0.000 |
4.343 |
0.000 |
| z |
0.000 |
0.000 |
1.680 |
|
| Polar |
|---|
| 3z2-r2 | 3.359 |
|---|
| x2-y2 | -6.910 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.747 |
0.000 |
0.000 |
| y |
0.000 |
8.882 |
0.000 |
| z |
0.000 |
0.000 |
8.115 |
<r2> (average value of r
2) Å
2
| <r2> |
88.210 |
| (<r2>)1/2 |
9.392 |