Jump to
S1C2
Energy calculated at B97D3/6-31G*
| | hartrees |
|---|
| Energy at 0K | -189.144638 |
| Energy at 298.15K | -189.151864 |
| HF Energy | -189.144638 |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B97D3/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3111 |
3050 |
2.19 |
|
|
|
| 2 |
A1 |
2970 |
2912 |
18.57 |
|
|
|
| 3 |
A1 |
1601 |
1570 |
14.94 |
|
|
|
| 4 |
A1 |
1470 |
1441 |
0.45 |
|
|
|
| 5 |
A1 |
1383 |
1356 |
19.29 |
|
|
|
| 6 |
A1 |
1079 |
1058 |
4.93 |
|
|
|
| 7 |
A1 |
821 |
805 |
0.06 |
|
|
|
| 8 |
A1 |
361 |
354 |
0.75 |
|
|
|
| 9 |
A2 |
3043 |
2983 |
0.00 |
|
|
|
| 10 |
A2 |
1477 |
1448 |
0.00 |
|
|
|
| 11 |
A2 |
1068 |
1047 |
0.00 |
|
|
|
| 12 |
A2 |
465 |
456 |
0.00 |
|
|
|
| 13 |
A2 |
54 |
53 |
0.00 |
|
|
|
| 14 |
B1 |
3036 |
2977 |
45.30 |
|
|
|
| 15 |
B1 |
1504 |
1474 |
18.76 |
|
|
|
| 16 |
B1 |
917 |
899 |
0.81 |
|
|
|
| 17 |
B1 |
181 |
177 |
0.00 |
|
|
|
| 18 |
B2 |
3110 |
3049 |
45.67 |
|
|
|
| 19 |
B2 |
2970 |
2911 |
2.65 |
|
|
|
| 20 |
B2 |
1459 |
1431 |
7.94 |
|
|
|
| 21 |
B2 |
1368 |
1341 |
2.87 |
|
|
|
| 22 |
B2 |
1159 |
1136 |
13.94 |
|
|
|
| 23 |
B2 |
888 |
870 |
21.76 |
|
|
|
| 24 |
B2 |
610 |
598 |
0.05 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 18052.3 cm
-1
Scaled (by 0.9804) Zero Point Vibrational Energy (zpe) 17698.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B97D3/6-31G*
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.624 |
-0.788 |
| N2 |
0.000 |
-0.624 |
-0.788 |
| C3 |
0.000 |
1.369 |
0.505 |
| C4 |
0.000 |
-1.369 |
0.505 |
| H5 |
0.000 |
2.439 |
0.270 |
| H6 |
0.000 |
-2.439 |
0.270 |
| H7 |
-0.892 |
1.128 |
1.106 |
| H8 |
0.892 |
1.128 |
1.106 |
| H9 |
0.892 |
-1.128 |
1.106 |
| H10 |
-0.892 |
-1.128 |
1.106 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| N1 | | 1.2489 | 1.4919 | 2.3761 | 2.1000 | 3.2406 | 2.1533 | 2.1533 | 2.7303 | 2.7303 |
N2 | 1.2489 | | 2.3761 | 1.4919 | 3.2406 | 2.1000 | 2.7303 | 2.7303 | 2.1533 | 2.1533 | C3 | 1.4919 | 2.3761 | | 2.7385 | 1.0950 | 3.8153 | 1.1023 | 1.1023 | 2.7193 | 2.7193 | C4 | 2.3761 | 1.4919 | 2.7385 | | 3.8153 | 1.0950 | 2.7193 | 2.7193 | 1.1023 | 1.1023 | H5 | 2.1000 | 3.2406 | 1.0950 | 3.8153 | | 4.8775 | 1.7924 | 1.7924 | 3.7707 | 3.7707 | H6 | 3.2406 | 2.1000 | 3.8153 | 1.0950 | 4.8775 | | 3.7707 | 3.7707 | 1.7924 | 1.7924 | H7 | 2.1533 | 2.7303 | 1.1023 | 2.7193 | 1.7924 | 3.7707 | | 1.7836 | 2.8763 | 2.2565 | H8 | 2.1533 | 2.7303 | 1.1023 | 2.7193 | 1.7924 | 3.7707 | 1.7836 | | 2.2565 | 2.8763 | H9 | 2.7303 | 2.1533 | 2.7193 | 1.1023 | 3.7707 | 1.7924 | 2.8763 | 2.2565 | | 1.7836 | H10 | 2.7303 | 2.1533 | 2.7193 | 1.1023 | 3.7707 | 1.7924 | 2.2565 | 2.8763 | 1.7836 | |
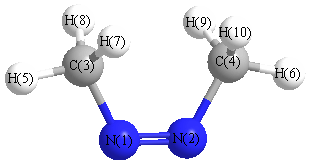 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
N2 |
C4 |
120.405 |
|
N1 |
C3 |
H5 |
107.558 |
| N1 |
C3 |
H7 |
111.443 |
|
N1 |
C3 |
H8 |
111.443 |
| N2 |
N1 |
C3 |
120.405 |
|
N2 |
C4 |
H6 |
107.558 |
| N2 |
C4 |
H9 |
111.443 |
|
N2 |
C4 |
H10 |
111.443 |
| H5 |
C3 |
H7 |
109.549 |
|
H5 |
C3 |
H8 |
109.549 |
| H6 |
C4 |
H9 |
109.549 |
|
H6 |
C4 |
H10 |
109.549 |
| H7 |
C3 |
H8 |
107.290 |
|
H9 |
C4 |
H10 |
107.290 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.152 |
|
|
|
| 2 |
N |
-0.152 |
|
|
|
| 3 |
C |
-0.367 |
|
|
|
| 4 |
C |
-0.367 |
|
|
|
| 5 |
H |
0.182 |
|
|
|
| 6 |
H |
0.182 |
|
|
|
| 7 |
H |
0.168 |
|
|
|
| 8 |
H |
0.168 |
|
|
|
| 9 |
H |
0.168 |
|
|
|
| 10 |
H |
0.168 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
3.118 |
3.118 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.300 |
0.000 |
0.000 |
| y |
0.000 |
-23.289 |
0.000 |
| z |
0.000 |
0.000 |
-28.459 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.573 |
0.000 |
0.000 |
| y |
0.000 |
3.091 |
0.000 |
| z |
0.000 |
0.000 |
-4.664 |
|
| Polar |
|---|
| 3z2-r2 | -9.329 |
|---|
| x2-y2 | -1.012 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.003 |
0.000 |
0.000 |
| y |
0.000 |
7.214 |
0.000 |
| z |
0.000 |
0.000 |
4.896 |
<r2> (average value of r
2) Å
2
| <r2> |
80.747 |
| (<r2>)1/2 |
8.986 |
Jump to
S1C1
Energy calculated at B97D3/6-31G*
| | hartrees |
|---|
| Energy at 0K | -189.144639 |
| Energy at 298.15K | -189.151844 |
| HF Energy | -189.144639 |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B97D3/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3112 |
3051 |
2.22 |
|
|
|
| 2 |
A |
3045 |
2985 |
0.00 |
|
|
|
| 3 |
A |
2972 |
2914 |
18.42 |
|
|
|
| 4 |
A |
1603 |
1571 |
15.10 |
|
|
|
| 5 |
A |
1477 |
1448 |
0.00 |
|
|
|
| 6 |
A |
1469 |
1441 |
0.42 |
|
|
|
| 7 |
A |
1383 |
1356 |
19.32 |
|
|
|
| 8 |
A |
1079 |
1058 |
4.91 |
|
|
|
| 9 |
A |
1068 |
1047 |
0.00 |
|
|
|
| 10 |
A |
819 |
803 |
0.06 |
|
|
|
| 11 |
A |
464 |
455 |
0.00 |
|
|
|
| 12 |
A |
361 |
354 |
0.75 |
|
|
|
| 13 |
A |
46 |
45 |
0.00 |
|
|
|
| 14 |
B |
3111 |
3050 |
45.45 |
|
|
|
| 15 |
B |
3039 |
2979 |
45.13 |
|
|
|
| 16 |
B |
2971 |
2913 |
2.69 |
|
|
|
| 17 |
B |
1504 |
1474 |
18.75 |
|
|
|
| 18 |
B |
1459 |
1430 |
8.01 |
|
|
|
| 19 |
B |
1367 |
1340 |
2.83 |
|
|
|
| 20 |
B |
1159 |
1136 |
14.01 |
|
|
|
| 21 |
B |
916 |
898 |
0.82 |
|
|
|
| 22 |
B |
885 |
868 |
21.94 |
|
|
|
| 23 |
B |
609 |
597 |
0.04 |
|
|
|
| 24 |
B |
179 |
175 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 18047.5 cm
-1
Scaled (by 0.9804) Zero Point Vibrational Energy (zpe) 17693.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B97D3/6-31G*
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
-0.000 |
0.624 |
-0.788 |
| N2 |
0.000 |
-0.624 |
-0.788 |
| C3 |
-0.000 |
1.370 |
0.505 |
| C4 |
0.000 |
-1.370 |
0.505 |
| H5 |
-0.001 |
2.439 |
0.269 |
| H6 |
0.001 |
-2.439 |
0.269 |
| H7 |
-0.892 |
1.128 |
1.107 |
| H8 |
0.892 |
1.129 |
1.106 |
| H9 |
0.892 |
-1.128 |
1.107 |
| H10 |
-0.892 |
-1.129 |
1.106 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| N1 | | 1.2486 | 1.4927 | 2.3767 | 2.1001 | 3.2405 | 2.1536 | 2.1534 | 2.7304 | 2.7307 |
N2 | 1.2486 | | 2.3767 | 1.4927 | 3.2405 | 2.1001 | 2.7304 | 2.7307 | 2.1536 | 2.1534 | C3 | 1.4927 | 2.3767 | | 2.7394 | 1.0949 | 3.8159 | 1.1021 | 1.1021 | 2.7194 | 2.7204 | C4 | 2.3767 | 1.4927 | 2.7394 | | 3.8159 | 1.0949 | 2.7194 | 2.7204 | 1.1021 | 1.1021 | H5 | 2.1001 | 3.2405 | 1.0949 | 3.8159 | | 4.8778 | 1.7925 | 1.7925 | 3.7710 | 3.7716 | H6 | 3.2405 | 2.1001 | 3.8159 | 1.0949 | 4.8778 | | 3.7710 | 3.7716 | 1.7925 | 1.7925 | H7 | 2.1536 | 2.7304 | 1.1021 | 2.7194 | 1.7925 | 3.7710 | | 1.7838 | 2.8757 | 2.2571 | H8 | 2.1534 | 2.7307 | 1.1021 | 2.7204 | 1.7925 | 3.7716 | 1.7838 | | 2.2571 | 2.8781 | H9 | 2.7304 | 2.1536 | 2.7194 | 1.1021 | 3.7710 | 1.7925 | 2.8757 | 2.2571 | | 1.7838 | H10 | 2.7307 | 2.1534 | 2.7204 | 1.1021 | 3.7716 | 1.7925 | 2.2571 | 2.8781 | 1.7838 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
N2 |
C4 |
121.246 |
|
N1 |
C3 |
H5 |
107.881 |
| N1 |
C3 |
H7 |
110.532 |
|
N1 |
C3 |
H8 |
112.602 |
| N2 |
N1 |
C3 |
121.246 |
|
N2 |
C4 |
H6 |
107.881 |
| N2 |
C4 |
H9 |
110.532 |
|
N2 |
C4 |
H10 |
112.602 |
| H5 |
C3 |
H7 |
108.714 |
|
H5 |
C3 |
H8 |
109.202 |
| H6 |
C4 |
H9 |
108.714 |
|
H6 |
C4 |
H10 |
109.202 |
| H7 |
C3 |
H8 |
107.844 |
|
H9 |
C4 |
H10 |
107.844 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.152 |
|
|
|
| 2 |
N |
-0.152 |
|
|
|
| 3 |
C |
-0.367 |
|
|
|
| 4 |
C |
-0.367 |
|
|
|
| 5 |
H |
0.182 |
|
|
|
| 6 |
H |
0.182 |
|
|
|
| 7 |
H |
0.168 |
|
|
|
| 8 |
H |
0.169 |
|
|
|
| 9 |
H |
0.168 |
|
|
|
| 10 |
H |
0.169 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
3.117 |
3.117 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.298 |
0.000 |
0.000 |
| y |
0.000 |
-23.289 |
0.000 |
| z |
0.000 |
0.000 |
-28.460 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.577 |
0.000 |
0.000 |
| y |
0.000 |
3.090 |
0.000 |
| z |
0.000 |
0.000 |
-4.667 |
|
| Polar |
|---|
| 3z2-r2 | -9.334 |
|---|
| x2-y2 | -1.009 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.003 |
0.000 |
0.000 |
| y |
0.000 |
7.216 |
0.000 |
| z |
0.000 |
0.000 |
4.896 |
<r2> (average value of r
2) Å
2
| <r2> |
80.772 |
| (<r2>)1/2 |
8.987 |