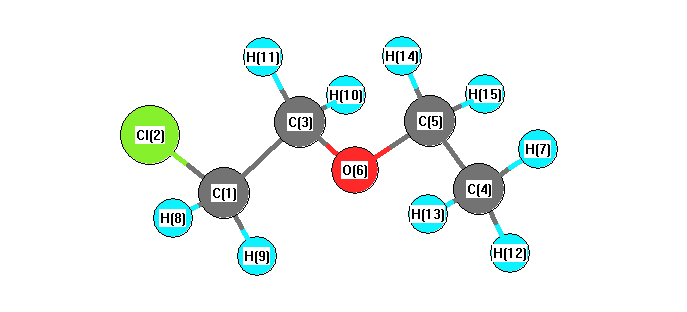
Vibrational Frequencies calculated at B97D3/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3082 |
3021 |
31.25 |
|
|
|
| 2 |
A' |
3057 |
2997 |
19.68 |
|
|
|
| 3 |
A' |
3004 |
2945 |
21.67 |
|
|
|
| 4 |
A' |
2933 |
2876 |
68.48 |
|
|
|
| 5 |
A' |
2913 |
2856 |
37.19 |
|
|
|
| 6 |
A' |
1536 |
1505 |
0.82 |
|
|
|
| 7 |
A' |
1516 |
1486 |
4.79 |
|
|
|
| 8 |
A' |
1502 |
1473 |
5.06 |
|
|
|
| 9 |
A' |
1491 |
1462 |
1.91 |
|
|
|
| 10 |
A' |
1437 |
1409 |
12.92 |
|
|
|
| 11 |
A' |
1401 |
1374 |
7.80 |
|
|
|
| 12 |
A' |
1373 |
1346 |
33.77 |
|
|
|
| 13 |
A' |
1276 |
1251 |
30.41 |
|
|
|
| 14 |
A' |
1143 |
1120 |
60.80 |
|
|
|
| 15 |
A' |
1115 |
1093 |
209.16 |
|
|
|
| 16 |
A' |
1054 |
1033 |
1.22 |
|
|
|
| 17 |
A' |
1019 |
999 |
14.19 |
|
|
|
| 18 |
A' |
885 |
868 |
15.90 |
|
|
|
| 19 |
A' |
726 |
712 |
49.84 |
|
|
|
| 20 |
A' |
472 |
462 |
0.35 |
|
|
|
| 21 |
A' |
370 |
363 |
2.45 |
|
|
|
| 22 |
A' |
261 |
256 |
3.54 |
|
|
|
| 23 |
A' |
119 |
116 |
1.33 |
|
|
|
| 24 |
A" |
3123 |
3062 |
13.05 |
|
|
|
| 25 |
A" |
3087 |
3027 |
33.58 |
|
|
|
| 26 |
A" |
2965 |
2907 |
47.26 |
|
|
|
| 27 |
A" |
2943 |
2886 |
80.88 |
|
|
|
| 28 |
A" |
1486 |
1457 |
5.03 |
|
|
|
| 29 |
A" |
1289 |
1263 |
1.47 |
|
|
|
| 30 |
A" |
1284 |
1259 |
0.59 |
|
|
|
| 31 |
A" |
1207 |
1183 |
5.06 |
|
|
|
| 32 |
A" |
1167 |
1145 |
3.42 |
|
|
|
| 33 |
A" |
1057 |
1036 |
2.52 |
|
|
|
| 34 |
A" |
818 |
802 |
0.82 |
|
|
|
| 35 |
A" |
799 |
783 |
0.32 |
|
|
|
| 36 |
A" |
249 |
244 |
0.83 |
|
|
|
| 37 |
A" |
147 |
144 |
7.42 |
|
|
|
| 38 |
A" |
71 |
69 |
1.77 |
|
|
|
| 39 |
A" |
50 |
49 |
0.49 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 27712.9 cm
-1
Scaled (by 0.9804) Zero Point Vibrational Energy (zpe) 27169.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.387 |
|
|
|
| 2 |
Cl |
-0.086 |
|
|
|
| 3 |
C |
-0.013 |
|
|
|
| 4 |
C |
-0.467 |
|
|
|
| 5 |
C |
-0.024 |
|
|
|
| 6 |
O |
-0.459 |
|
|
|
| 7 |
H |
0.152 |
|
|
|
| 8 |
H |
0.207 |
|
|
|
| 9 |
H |
0.207 |
|
|
|
| 10 |
H |
0.143 |
|
|
|
| 11 |
H |
0.143 |
|
|
|
| 12 |
H |
0.165 |
|
|
|
| 13 |
H |
0.165 |
|
|
|
| 14 |
H |
0.128 |
|
|
|
| 15 |
H |
0.128 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.142 |
-0.370 |
0.000 |
2.174 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-45.942 |
3.183 |
0.000 |
| y |
3.183 |
-45.437 |
0.000 |
| z |
0.000 |
0.000 |
-43.861 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.293 |
3.183 |
0.000 |
| y |
3.183 |
-0.535 |
0.000 |
| z |
0.000 |
0.000 |
1.828 |
|
| Polar |
|---|
| 3z2-r2 | 3.657 |
|---|
| x2-y2 | -0.506 |
|---|
| xy | 3.183 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.157 |
-1.548 |
0.000 |
| y |
-1.548 |
8.102 |
0.000 |
| z |
0.000 |
0.000 |
7.012 |
<r2> (average value of r
2) Å
2
| <r2> |
369.591 |
| (<r2>)1/2 |
19.225 |