Jump to
S1C2
Energy calculated at B97D3/6-31G*
| | hartrees |
|---|
| Energy at 0K | -342.194610 |
| Energy at 298.15K | |
| HF Energy | -342.194610 |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B97D3/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3027 |
2968 |
52.99 |
|
|
|
| 2 |
A1 |
1885 |
1848 |
522.34 |
|
|
|
| 3 |
A1 |
1534 |
1504 |
2.25 |
|
|
|
| 4 |
A1 |
1368 |
1341 |
0.81 |
|
|
|
| 5 |
A1 |
1074 |
1053 |
199.95 |
|
|
|
| 6 |
A1 |
950 |
931 |
19.91 |
|
|
|
| 7 |
A1 |
859 |
842 |
2.19 |
|
|
|
| 8 |
A1 |
705 |
692 |
3.46 |
|
|
|
| 9 |
A2 |
3061 |
3001 |
0.00 |
|
|
|
| 10 |
A2 |
1224 |
1200 |
0.00 |
|
|
|
| 11 |
A2 |
1140 |
1118 |
0.00 |
|
|
|
| 12 |
A2 |
121i |
119i |
0.00 |
|
|
|
| 13 |
B1 |
3083 |
3022 |
51.76 |
|
|
|
| 14 |
B1 |
1222 |
1198 |
0.56 |
|
|
|
| 15 |
B1 |
848 |
831 |
0.36 |
|
|
|
| 16 |
B1 |
720 |
706 |
17.88 |
|
|
|
| 17 |
B1 |
179 |
176 |
1.70 |
|
|
|
| 18 |
B2 |
3021 |
2962 |
32.20 |
|
|
|
| 19 |
B2 |
1521 |
1492 |
2.23 |
|
|
|
| 20 |
B2 |
1392 |
1365 |
39.49 |
|
|
|
| 21 |
B2 |
1099 |
1078 |
272.50 |
|
|
|
| 22 |
B2 |
1038 |
1018 |
8.60 |
|
|
|
| 23 |
B2 |
730 |
716 |
0.00 |
|
|
|
| 24 |
B2 |
502 |
492 |
0.22 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16030.2 cm
-1
Scaled (by 0.9804) Zero Point Vibrational Energy (zpe) 15716.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B97D3/6-31G*
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.859 |
| O2 |
0.000 |
0.000 |
2.060 |
| O3 |
0.000 |
1.128 |
0.079 |
| O4 |
0.000 |
-1.128 |
0.079 |
| C5 |
0.000 |
0.775 |
-1.314 |
| C6 |
0.000 |
-0.775 |
-1.314 |
| H7 |
-0.895 |
1.203 |
-1.784 |
| H8 |
0.895 |
1.203 |
-1.784 |
| H9 |
0.895 |
-1.203 |
-1.784 |
| H10 |
-0.895 |
-1.203 |
-1.784 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.2010 | 1.3714 | 1.3714 | 2.3076 | 2.3076 | 3.0388 | 3.0388 | 3.0388 | 3.0388 |
O2 | 1.2010 | | 2.2798 | 2.2798 | 3.4624 | 3.4624 | 4.1261 | 4.1261 | 4.1261 | 4.1261 | O3 | 1.3714 | 2.2798 | | 2.2554 | 1.4371 | 2.3584 | 2.0680 | 2.0680 | 3.1151 | 3.1151 | O4 | 1.3714 | 2.2798 | 2.2554 | | 2.3584 | 1.4371 | 3.1151 | 3.1151 | 2.0680 | 2.0680 | C5 | 2.3076 | 3.4624 | 1.4371 | 2.3584 | | 1.5504 | 1.0979 | 1.0979 | 2.2217 | 2.2217 | C6 | 2.3076 | 3.4624 | 2.3584 | 1.4371 | 1.5504 | | 2.2217 | 2.2217 | 1.0979 | 1.0979 | H7 | 3.0388 | 4.1261 | 2.0680 | 3.1151 | 1.0979 | 2.2217 | | 1.7909 | 2.9996 | 2.4062 | H8 | 3.0388 | 4.1261 | 2.0680 | 3.1151 | 1.0979 | 2.2217 | 1.7909 | | 2.4062 | 2.9996 | H9 | 3.0388 | 4.1261 | 3.1151 | 2.0680 | 2.2217 | 1.0979 | 2.9996 | 2.4062 | | 1.7909 | H10 | 3.0388 | 4.1261 | 3.1151 | 2.0680 | 2.2217 | 1.0979 | 2.4062 | 2.9996 | 1.7909 | |
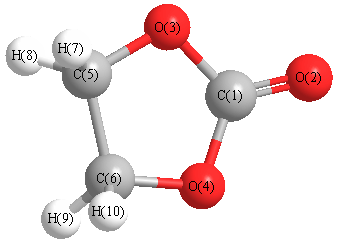 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
111.526 |
|
C1 |
O4 |
C6 |
111.526 |
| O2 |
C1 |
O3 |
125.470 |
|
O2 |
C1 |
O4 |
125.470 |
| O3 |
C1 |
O4 |
109.060 |
|
O3 |
C5 |
C6 |
103.944 |
| O3 |
C5 |
H7 |
110.004 |
|
O3 |
C5 |
H8 |
110.004 |
| O4 |
C6 |
C5 |
103.944 |
|
O4 |
C6 |
H9 |
110.004 |
| O4 |
C6 |
H10 |
110.004 |
|
C5 |
C6 |
H9 |
112.247 |
| C5 |
C6 |
H10 |
112.247 |
|
C6 |
C5 |
H7 |
112.247 |
| C6 |
C5 |
H8 |
112.247 |
|
H7 |
C5 |
H8 |
108.339 |
| H9 |
C6 |
H10 |
108.339 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.761 |
|
|
|
| 2 |
O |
-0.440 |
|
|
|
| 3 |
O |
-0.434 |
|
|
|
| 4 |
O |
-0.434 |
|
|
|
| 5 |
C |
-0.081 |
|
|
|
| 6 |
C |
-0.081 |
|
|
|
| 7 |
H |
0.177 |
|
|
|
| 8 |
H |
0.177 |
|
|
|
| 9 |
H |
0.177 |
|
|
|
| 10 |
H |
0.177 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-5.189 |
5.189 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.678 |
0.000 |
0.000 |
| y |
0.000 |
-35.463 |
0.000 |
| z |
0.000 |
0.000 |
-35.164 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.636 |
0.000 |
0.000 |
| y |
0.000 |
-2.042 |
0.000 |
| z |
0.000 |
0.000 |
-1.594 |
|
| Polar |
|---|
| 3z2-r2 | -3.188 |
|---|
| x2-y2 | 3.785 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.212 |
0.000 |
0.000 |
| y |
0.000 |
5.183 |
0.000 |
| z |
0.000 |
0.000 |
7.550 |
<r2> (average value of r
2) Å
2
| <r2> |
129.795 |
| (<r2>)1/2 |
11.393 |
Jump to
S1C1
Energy calculated at B97D3/6-31G*
| | hartrees |
|---|
| Energy at 0K | -342.195236 |
| Energy at 298.15K | -342.201603 |
| HF Energy | -342.195236 |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B97D3/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3085 |
3024 |
15.43 |
|
|
|
| 2 |
A |
3012 |
2953 |
30.91 |
|
|
|
| 3 |
A |
1887 |
1850 |
509.97 |
|
|
|
| 4 |
A |
1522 |
1492 |
2.17 |
|
|
|
| 5 |
A |
1372 |
1345 |
3.78 |
|
|
|
| 6 |
A |
1228 |
1204 |
10.43 |
|
|
|
| 7 |
A |
1141 |
1119 |
1.63 |
|
|
|
| 8 |
A |
1060 |
1040 |
182.76 |
|
|
|
| 9 |
A |
947 |
928 |
18.74 |
|
|
|
| 10 |
A |
860 |
843 |
4.33 |
|
|
|
| 11 |
A |
703 |
689 |
3.67 |
|
|
|
| 12 |
A |
158 |
155 |
0.40 |
|
|
|
| 13 |
B |
3097 |
3036 |
36.28 |
|
|
|
| 14 |
B |
3016 |
2957 |
49.30 |
|
|
|
| 15 |
B |
1514 |
1484 |
3.07 |
|
|
|
| 16 |
B |
1384 |
1357 |
33.58 |
|
|
|
| 17 |
B |
1220 |
1196 |
4.14 |
|
|
|
| 18 |
B |
1088 |
1066 |
261.47 |
|
|
|
| 19 |
B |
1028 |
1008 |
5.09 |
|
|
|
| 20 |
B |
877 |
860 |
1.31 |
|
|
|
| 21 |
B |
730 |
716 |
18.23 |
|
|
|
| 22 |
B |
675 |
662 |
0.73 |
|
|
|
| 23 |
B |
502 |
493 |
0.17 |
|
|
|
| 24 |
B |
185 |
182 |
1.52 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16145.5 cm
-1
Scaled (by 0.9804) Zero Point Vibrational Energy (zpe) 15829.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B97D3/6-31G*
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.855 |
| O2 |
0.000 |
0.000 |
2.055 |
| O3 |
0.000 |
1.128 |
0.069 |
| O4 |
0.000 |
-1.128 |
0.069 |
| C5 |
0.195 |
0.743 |
-1.305 |
| C6 |
-0.195 |
-0.743 |
-1.305 |
| H7 |
-0.445 |
1.368 |
-1.938 |
| H8 |
1.250 |
0.903 |
-1.571 |
| H9 |
0.445 |
-1.368 |
-1.938 |
| H10 |
-1.250 |
-0.903 |
-1.571 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.2007 | 1.3744 | 1.3744 | 2.2927 | 2.2927 | 3.1419 | 2.8749 | 3.1419 | 2.8749 |
O2 | 1.2007 | | 2.2840 | 2.2840 | 3.4476 | 3.4476 | 4.2452 | 3.9412 | 4.2452 | 3.9412 | O3 | 1.3744 | 2.2840 | | 2.2560 | 1.4409 | 2.3302 | 2.0706 | 2.0753 | 3.2342 | 2.8948 | O4 | 1.3744 | 2.2840 | 2.2560 | | 2.3302 | 1.4409 | 3.2342 | 2.8948 | 2.0706 | 2.0753 | C5 | 2.2927 | 3.4476 | 1.4409 | 2.3302 | | 1.5368 | 1.0958 | 1.1001 | 2.2183 | 2.2065 | C6 | 2.2927 | 3.4476 | 2.3302 | 1.4409 | 1.5368 | | 2.2183 | 2.2065 | 1.0958 | 1.1001 | H7 | 3.1419 | 4.2452 | 2.0706 | 3.2342 | 1.0958 | 2.2183 | | 1.7960 | 2.8772 | 2.4371 | H8 | 2.8749 | 3.9412 | 2.0753 | 2.8948 | 1.1001 | 2.2065 | 1.7960 | | 2.4371 | 3.0843 | H9 | 3.1419 | 4.2452 | 3.2342 | 2.0706 | 2.2183 | 1.0958 | 2.8772 | 2.4371 | | 1.7960 | H10 | 2.8749 | 3.9412 | 2.8948 | 2.0753 | 2.2065 | 1.1001 | 2.4371 | 3.0843 | 1.7960 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
108.831 |
|
C1 |
O4 |
C6 |
108.831 |
| O2 |
C1 |
O3 |
125.370 |
|
O2 |
C1 |
O4 |
125.370 |
| O3 |
C1 |
O4 |
109.261 |
|
O3 |
C5 |
C6 |
102.659 |
| O3 |
C5 |
H7 |
109.496 |
|
O3 |
C5 |
H8 |
109.465 |
| O4 |
C6 |
C5 |
102.659 |
|
O4 |
C6 |
H9 |
109.496 |
| O4 |
C6 |
H10 |
109.465 |
|
C5 |
C6 |
H9 |
113.714 |
| C5 |
C6 |
H10 |
111.751 |
|
C6 |
C5 |
H7 |
113.714 |
| C6 |
C5 |
H8 |
111.751 |
|
H7 |
C5 |
H8 |
109.506 |
| H9 |
C6 |
H10 |
109.506 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.766 |
|
|
|
| 2 |
O |
-0.441 |
|
|
|
| 3 |
O |
-0.435 |
|
|
|
| 4 |
O |
-0.435 |
|
|
|
| 5 |
C |
-0.083 |
|
|
|
| 6 |
C |
-0.083 |
|
|
|
| 7 |
H |
0.180 |
|
|
|
| 8 |
H |
0.176 |
|
|
|
| 9 |
H |
0.180 |
|
|
|
| 10 |
H |
0.176 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-5.133 |
5.133 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.707 |
0.242 |
0.000 |
| y |
0.242 |
-35.504 |
0.000 |
| z |
0.000 |
0.000 |
-35.016 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.553 |
0.242 |
0.000 |
| y |
0.242 |
-2.142 |
0.000 |
| z |
0.000 |
0.000 |
-1.411 |
|
| Polar |
|---|
| 3z2-r2 | -2.821 |
|---|
| x2-y2 | 3.797 |
|---|
| xy | 0.242 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.285 |
0.136 |
0.000 |
| y |
0.136 |
5.133 |
0.000 |
| z |
0.000 |
0.000 |
7.528 |
<r2> (average value of r
2) Å
2
| <r2> |
128.786 |
| (<r2>)1/2 |
11.348 |