Jump to
S1C2
Energy calculated at B97D3/6-311+G(3df,2p)
| | hartrees |
|---|
| Energy at 0K | -271.711173 |
| Energy at 298.15K | |
| HF Energy | -271.711173 |
| Nuclear repulsion energy | 235.131333 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B97D3/6-311+G(3df,2p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3090 |
3050 |
14.59 |
|
|
|
| 2 |
A' |
3039 |
2999 |
45.21 |
|
|
|
| 3 |
A' |
2989 |
2950 |
21.00 |
|
|
|
| 4 |
A' |
2975 |
2936 |
8.67 |
|
|
|
| 5 |
A' |
2964 |
2926 |
34.54 |
|
|
|
| 6 |
A' |
2943 |
2905 |
16.92 |
|
|
|
| 7 |
A' |
1732 |
1710 |
158.33 |
|
|
|
| 8 |
A' |
1478 |
1459 |
6.15 |
|
|
|
| 9 |
A' |
1462 |
1443 |
2.02 |
|
|
|
| 10 |
A' |
1437 |
1418 |
20.85 |
|
|
|
| 11 |
A' |
1417 |
1398 |
5.49 |
|
|
|
| 12 |
A' |
1382 |
1364 |
0.68 |
|
|
|
| 13 |
A' |
1358 |
1340 |
30.87 |
|
|
|
| 14 |
A' |
1348 |
1330 |
29.42 |
|
|
|
| 15 |
A' |
1291 |
1275 |
15.15 |
|
|
|
| 16 |
A' |
1151 |
1136 |
73.31 |
|
|
|
| 17 |
A' |
1103 |
1089 |
0.28 |
|
|
|
| 18 |
A' |
1026 |
1013 |
0.17 |
|
|
|
| 19 |
A' |
936 |
923 |
6.34 |
|
|
|
| 20 |
A' |
883 |
872 |
15.03 |
|
|
|
| 21 |
A' |
798 |
788 |
3.15 |
|
|
|
| 22 |
A' |
580 |
573 |
8.90 |
|
|
|
| 23 |
A' |
390 |
385 |
1.31 |
|
|
|
| 24 |
A' |
334 |
329 |
2.08 |
|
|
|
| 25 |
A' |
167 |
165 |
4.70 |
|
|
|
| 26 |
A" |
3038 |
2998 |
73.47 |
|
|
|
| 27 |
A" |
3033 |
2993 |
4.74 |
|
|
|
| 28 |
A" |
3011 |
2972 |
0.90 |
|
|
|
| 29 |
A" |
2968 |
2929 |
8.16 |
|
|
|
| 30 |
A" |
1472 |
1453 |
6.27 |
|
|
|
| 31 |
A" |
1446 |
1427 |
10.00 |
|
|
|
| 32 |
A" |
1298 |
1281 |
0.08 |
|
|
|
| 33 |
A" |
1226 |
1210 |
0.03 |
|
|
|
| 34 |
A" |
1112 |
1097 |
0.47 |
|
|
|
| 35 |
A" |
953 |
941 |
0.73 |
|
|
|
| 36 |
A" |
813 |
802 |
0.50 |
|
|
|
| 37 |
A" |
707 |
698 |
3.10 |
|
|
|
| 38 |
A" |
462 |
456 |
0.04 |
|
|
|
| 39 |
A" |
238 |
235 |
0.03 |
|
|
|
| 40 |
A" |
79 |
78 |
0.48 |
|
|
|
| 41 |
A" |
63 |
62 |
0.73 |
|
|
|
| 42 |
A" |
76i |
75i |
0.19 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30056.9 cm
-1
Scaled (by 0.987) Zero Point Vibrational Energy (zpe) 29666.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B97D3/6-311+G(3df,2p)
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-2.407 |
1.377 |
0.000 |
| C2 |
-1.472 |
0.164 |
0.000 |
| C3 |
0.000 |
0.569 |
0.000 |
| C4 |
0.985 |
-0.594 |
0.000 |
| C5 |
2.457 |
-0.215 |
0.000 |
| O6 |
0.623 |
-1.754 |
0.000 |
| H7 |
-3.457 |
1.068 |
0.000 |
| H8 |
-2.244 |
2.004 |
0.884 |
| H9 |
-2.244 |
2.004 |
-0.884 |
| H10 |
-1.669 |
-0.467 |
0.873 |
| H11 |
-1.669 |
-0.467 |
-0.873 |
| H12 |
0.233 |
1.201 |
-0.871 |
| H13 |
0.233 |
1.201 |
0.871 |
| H14 |
3.079 |
-1.111 |
0.000 |
| H15 |
2.690 |
0.397 |
-0.879 |
| H16 |
2.690 |
0.397 |
0.879 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
| C1 | | 1.5307 | 2.5388 | 3.9230 | 5.1175 | 4.3569 | 1.0948 | 1.0965 | 1.0965 | 2.1689 | 2.1689 | 2.7850 | 2.7850 | 6.0232 | 5.2638 | 5.2638 |
C2 | 1.5307 | | 1.5267 | 2.5718 | 3.9473 | 2.8410 | 2.1811 | 2.1823 | 2.1823 | 1.0955 | 1.0955 | 2.1768 | 2.1768 | 4.7263 | 4.2602 | 4.2602 | C3 | 2.5388 | 1.5267 | | 1.5243 | 2.5788 | 2.4053 | 3.4932 | 2.8068 | 2.8068 | 2.1499 | 2.1499 | 1.1007 | 1.1007 | 3.5071 | 2.8350 | 2.8350 | C4 | 3.9230 | 2.5718 | 1.5243 | | 1.5199 | 1.2153 | 4.7433 | 4.2382 | 4.2382 | 2.7975 | 2.7975 | 2.1323 | 2.1323 | 2.1564 | 2.1593 | 2.1593 | C5 | 5.1175 | 3.9473 | 2.5788 | 1.5199 | | 2.3946 | 6.0517 | 5.2730 | 5.2730 | 4.2253 | 4.2253 | 2.7765 | 2.7765 | 1.0907 | 1.0965 | 1.0965 | O6 | 4.3569 | 2.8410 | 2.4053 | 1.2153 | 2.3946 | | 4.9614 | 4.8092 | 4.8092 | 2.7706 | 2.7706 | 3.1054 | 3.1054 | 2.5388 | 3.1104 | 3.1104 | H7 | 1.0948 | 2.1811 | 3.4932 | 4.7433 | 6.0517 | 4.9614 | | 1.7692 | 1.7692 | 2.5129 | 2.5129 | 3.7936 | 3.7936 | 6.8897 | 6.2458 | 6.2458 | H8 | 1.0965 | 2.1823 | 2.8068 | 4.2382 | 5.2730 | 4.8092 | 1.7692 | | 1.7686 | 2.5367 | 3.0862 | 3.1401 | 2.6038 | 6.2304 | 5.4805 | 5.1889 | H9 | 1.0965 | 2.1823 | 2.8068 | 4.2382 | 5.2730 | 4.8092 | 1.7692 | 1.7686 | | 3.0862 | 2.5367 | 2.6038 | 3.1401 | 6.2304 | 5.1889 | 5.4805 | H10 | 2.1689 | 1.0955 | 2.1499 | 2.7975 | 4.2253 | 2.7706 | 2.5129 | 2.5367 | 3.0862 | | 1.7469 | 3.0725 | 2.5294 | 4.8707 | 4.7771 | 4.4439 | H11 | 2.1689 | 1.0955 | 2.1499 | 2.7975 | 4.2253 | 2.7706 | 2.5129 | 3.0862 | 2.5367 | 1.7469 | | 2.5294 | 3.0725 | 4.8707 | 4.4439 | 4.7771 | H12 | 2.7850 | 2.1768 | 1.1007 | 2.1323 | 2.7765 | 3.1054 | 3.7936 | 3.1401 | 2.6038 | 3.0725 | 2.5294 | | 1.7415 | 3.7685 | 2.5851 | 3.1218 | H13 | 2.7850 | 2.1768 | 1.1007 | 2.1323 | 2.7765 | 3.1054 | 3.7936 | 2.6038 | 3.1401 | 2.5294 | 3.0725 | 1.7415 | | 3.7685 | 3.1218 | 2.5851 | H14 | 6.0232 | 4.7263 | 3.5071 | 2.1564 | 1.0907 | 2.5388 | 6.8897 | 6.2304 | 6.2304 | 4.8707 | 4.8707 | 3.7685 | 3.7685 | | 1.7886 | 1.7886 | H15 | 5.2638 | 4.2602 | 2.8350 | 2.1593 | 1.0965 | 3.1104 | 6.2458 | 5.4805 | 5.1889 | 4.7771 | 4.4439 | 2.5851 | 3.1218 | 1.7886 | | 1.7589 | H16 | 5.2638 | 4.2602 | 2.8350 | 2.1593 | 1.0965 | 3.1104 | 6.2458 | 5.1889 | 5.4805 | 4.4439 | 4.7771 | 3.1218 | 2.5851 | 1.7886 | 1.7589 | |
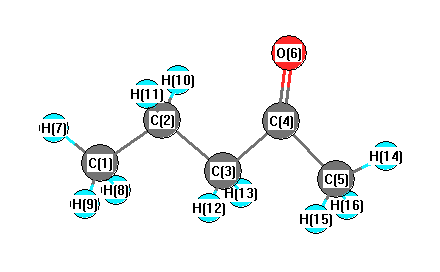 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
112.602 |
|
C1 |
C2 |
H10 |
110.266 |
| C1 |
C2 |
H11 |
110.266 |
|
C2 |
C1 |
H7 |
111.713 |
| C2 |
C1 |
H8 |
111.450 |
|
C2 |
C1 |
H9 |
111.450 |
| C2 |
C3 |
C4 |
115.367 |
|
C2 |
C3 |
H12 |
110.513 |
| C2 |
C3 |
H13 |
110.513 |
|
C3 |
C2 |
H10 |
109.066 |
| C3 |
C2 |
H11 |
109.066 |
|
C3 |
C4 |
C5 |
115.610 |
| C3 |
C4 |
O6 |
122.322 |
|
C4 |
C3 |
H12 |
107.571 |
| C4 |
C3 |
H13 |
107.571 |
|
C4 |
C5 |
H14 |
111.094 |
| C4 |
C5 |
H15 |
110.428 |
|
C4 |
C5 |
H16 |
110.428 |
| C5 |
C4 |
O6 |
122.068 |
|
H7 |
C1 |
H8 |
107.370 |
| H7 |
C1 |
H9 |
107.370 |
|
H8 |
C1 |
H9 |
107.243 |
| H10 |
C2 |
H11 |
105.306 |
|
H12 |
C3 |
H13 |
104.711 |
| H14 |
C5 |
H15 |
109.009 |
|
H14 |
C5 |
H16 |
109.009 |
| H15 |
C5 |
H16 |
106.759 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/6-311+G(3df,2p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.478 |
|
|
|
| 2 |
C |
0.065 |
|
|
|
| 3 |
C |
-0.315 |
|
|
|
| 4 |
C |
0.486 |
|
|
|
| 5 |
C |
-0.343 |
|
|
|
| 6 |
O |
-0.714 |
|
|
|
| 7 |
H |
0.116 |
|
|
|
| 8 |
H |
0.125 |
|
|
|
| 9 |
H |
0.125 |
|
|
|
| 10 |
H |
0.118 |
|
|
|
| 11 |
H |
0.118 |
|
|
|
| 12 |
H |
0.141 |
|
|
|
| 13 |
H |
0.141 |
|
|
|
| 14 |
H |
0.128 |
|
|
|
| 15 |
H |
0.143 |
|
|
|
| 16 |
H |
0.143 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.664 |
2.674 |
0.000 |
2.756 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.836 |
1.498 |
0.000 |
| y |
1.498 |
-44.774 |
0.000 |
| z |
0.000 |
0.000 |
-37.420 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.261 |
1.498 |
0.000 |
| y |
1.498 |
-8.146 |
0.000 |
| z |
0.000 |
0.000 |
2.885 |
|
| Polar |
|---|
| 3z2-r2 | 5.771 |
|---|
| x2-y2 | 8.938 |
|---|
| xy | 1.498 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.990 |
-0.601 |
0.000 |
| y |
-0.601 |
10.445 |
0.000 |
| z |
0.000 |
0.000 |
8.118 |
<r2> (average value of r
2) Å
2
| <r2> |
229.824 |
| (<r2>)1/2 |
15.160 |
Jump to
S1C1
Energy calculated at B97D3/6-311+G(3df,2p)
| | hartrees |
|---|
| Energy at 0K | -271.711173 |
| Energy at 298.15K | |
| HF Energy | -271.711173 |
| Nuclear repulsion energy | 235.130972 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B97D3/6-311+G(3df,2p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3090 |
3050 |
14.58 |
|
|
|
| 2 |
A' |
3039 |
2999 |
45.17 |
|
|
|
| 3 |
A' |
2989 |
2950 |
21.02 |
|
|
|
| 4 |
A' |
2974 |
2936 |
8.67 |
|
|
|
| 5 |
A' |
2964 |
2926 |
34.51 |
|
|
|
| 6 |
A' |
2942 |
2904 |
16.97 |
|
|
|
| 7 |
A' |
1733 |
1710 |
158.36 |
|
|
|
| 8 |
A' |
1478 |
1459 |
6.15 |
|
|
|
| 9 |
A' |
1462 |
1443 |
2.01 |
|
|
|
| 10 |
A' |
1437 |
1418 |
20.85 |
|
|
|
| 11 |
A' |
1417 |
1398 |
5.52 |
|
|
|
| 12 |
A' |
1382 |
1364 |
0.69 |
|
|
|
| 13 |
A' |
1358 |
1340 |
31.13 |
|
|
|
| 14 |
A' |
1348 |
1330 |
29.28 |
|
|
|
| 15 |
A' |
1292 |
1275 |
14.99 |
|
|
|
| 16 |
A' |
1151 |
1136 |
73.32 |
|
|
|
| 17 |
A' |
1103 |
1089 |
0.29 |
|
|
|
| 18 |
A' |
1026 |
1012 |
0.16 |
|
|
|
| 19 |
A' |
936 |
923 |
6.31 |
|
|
|
| 20 |
A' |
883 |
872 |
15.14 |
|
|
|
| 21 |
A' |
799 |
788 |
3.11 |
|
|
|
| 22 |
A' |
580 |
573 |
8.88 |
|
|
|
| 23 |
A' |
390 |
385 |
1.31 |
|
|
|
| 24 |
A' |
334 |
329 |
2.08 |
|
|
|
| 25 |
A' |
167 |
165 |
4.70 |
|
|
|
| 26 |
A" |
3038 |
2998 |
72.92 |
|
|
|
| 27 |
A" |
3032 |
2993 |
5.38 |
|
|
|
| 28 |
A" |
3011 |
2972 |
0.78 |
|
|
|
| 29 |
A" |
2967 |
2928 |
8.19 |
|
|
|
| 30 |
A" |
1472 |
1453 |
6.27 |
|
|
|
| 31 |
A" |
1446 |
1427 |
9.99 |
|
|
|
| 32 |
A" |
1298 |
1281 |
0.08 |
|
|
|
| 33 |
A" |
1226 |
1210 |
0.03 |
|
|
|
| 34 |
A" |
1112 |
1098 |
0.47 |
|
|
|
| 35 |
A" |
954 |
941 |
0.74 |
|
|
|
| 36 |
A" |
813 |
802 |
0.50 |
|
|
|
| 37 |
A" |
707 |
698 |
3.10 |
|
|
|
| 38 |
A" |
462 |
456 |
0.04 |
|
|
|
| 39 |
A" |
238 |
235 |
0.03 |
|
|
|
| 40 |
A" |
80 |
79 |
0.49 |
|
|
|
| 41 |
A" |
64 |
63 |
0.72 |
|
|
|
| 42 |
A" |
75i |
74i |
0.18 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30057.6 cm
-1
Scaled (by 0.987) Zero Point Vibrational Energy (zpe) 29666.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B97D3/6-311+G(3df,2p)
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-2.406 |
1.378 |
0.000 |
| C2 |
-1.472 |
0.165 |
0.000 |
| C3 |
0.000 |
0.568 |
0.000 |
| C4 |
0.985 |
-0.595 |
0.000 |
| C5 |
2.457 |
-0.215 |
0.000 |
| O6 |
0.623 |
-1.755 |
0.000 |
| H7 |
-3.457 |
1.070 |
0.000 |
| H8 |
-2.243 |
2.005 |
0.884 |
| H9 |
-2.243 |
2.005 |
-0.884 |
| H10 |
-1.670 |
-0.466 |
0.874 |
| H11 |
-1.670 |
-0.466 |
-0.874 |
| H12 |
0.233 |
1.200 |
-0.871 |
| H13 |
0.233 |
1.200 |
0.871 |
| H14 |
3.078 |
-1.112 |
0.000 |
| H15 |
2.690 |
0.397 |
-0.879 |
| H16 |
2.690 |
0.397 |
0.879 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
| C1 | | 1.5308 | 2.5387 | 3.9229 | 5.1172 | 4.3574 | 1.0948 | 1.0965 | 1.0965 | 2.1690 | 2.1690 | 2.7844 | 2.7844 | 6.0229 | 5.2638 | 5.2638 |
C2 | 1.5308 | | 1.5267 | 2.5719 | 3.9475 | 2.8415 | 2.1813 | 2.1824 | 2.1824 | 1.0955 | 1.0955 | 2.1766 | 2.1766 | 4.7263 | 4.2607 | 4.2607 | C3 | 2.5387 | 1.5267 | | 1.5239 | 2.5787 | 2.4050 | 3.4931 | 2.8066 | 2.8066 | 2.1500 | 2.1500 | 1.1008 | 1.1008 | 3.5068 | 2.8354 | 2.8354 | C4 | 3.9229 | 2.5719 | 1.5239 | | 1.5201 | 1.2152 | 4.7434 | 4.2378 | 4.2378 | 2.7981 | 2.7981 | 2.1321 | 2.1321 | 2.1563 | 2.1598 | 2.1598 | C5 | 5.1172 | 3.9475 | 2.5787 | 1.5201 | | 2.3945 | 6.0517 | 5.2724 | 5.2724 | 4.2259 | 4.2259 | 2.7764 | 2.7764 | 1.0907 | 1.0965 | 1.0965 | O6 | 4.3574 | 2.8415 | 2.4050 | 1.2152 | 2.3945 | | 4.9622 | 4.8094 | 4.8094 | 2.7716 | 2.7716 | 3.1052 | 3.1052 | 2.5384 | 3.1107 | 3.1107 | H7 | 1.0948 | 2.1813 | 3.4931 | 4.7434 | 6.0517 | 4.9622 | | 1.7692 | 1.7692 | 2.5129 | 2.5129 | 3.7931 | 3.7931 | 6.8897 | 6.2460 | 6.2460 | H8 | 1.0965 | 2.1824 | 2.8066 | 4.2378 | 5.2724 | 4.8094 | 1.7692 | | 1.7687 | 2.5367 | 3.0862 | 3.1394 | 2.6030 | 6.2297 | 5.4801 | 5.1885 | H9 | 1.0965 | 2.1824 | 2.8066 | 4.2378 | 5.2724 | 4.8094 | 1.7692 | 1.7687 | | 3.0862 | 2.5367 | 2.6030 | 3.1394 | 6.2297 | 5.1885 | 5.4801 | H10 | 2.1690 | 1.0955 | 2.1500 | 2.7981 | 4.2259 | 2.7716 | 2.5129 | 2.5367 | 3.0862 | | 1.7471 | 3.0725 | 2.5294 | 4.8712 | 4.7782 | 4.4450 | H11 | 2.1690 | 1.0955 | 2.1500 | 2.7981 | 4.2259 | 2.7716 | 2.5129 | 3.0862 | 2.5367 | 1.7471 | | 2.5294 | 3.0725 | 4.8712 | 4.4450 | 4.7782 | H12 | 2.7844 | 2.1766 | 1.1008 | 2.1321 | 2.7764 | 3.1052 | 3.7931 | 3.1394 | 2.6030 | 3.0725 | 2.5294 | | 1.7415 | 3.7683 | 2.5856 | 3.1222 | H13 | 2.7844 | 2.1766 | 1.1008 | 2.1321 | 2.7764 | 3.1052 | 3.7931 | 2.6030 | 3.1394 | 2.5294 | 3.0725 | 1.7415 | | 3.7683 | 3.1222 | 2.5856 | H14 | 6.0229 | 4.7263 | 3.5068 | 2.1563 | 1.0907 | 2.5384 | 6.8897 | 6.2297 | 6.2297 | 4.8712 | 4.8712 | 3.7683 | 3.7683 | | 1.7886 | 1.7886 | H15 | 5.2638 | 4.2607 | 2.8354 | 2.1598 | 1.0965 | 3.1107 | 6.2460 | 5.4801 | 5.1885 | 4.7782 | 4.4450 | 2.5856 | 3.1222 | 1.7886 | | 1.7589 | H16 | 5.2638 | 4.2607 | 2.8354 | 2.1598 | 1.0965 | 3.1107 | 6.2460 | 5.1885 | 5.4801 | 4.4450 | 4.7782 | 3.1222 | 2.5856 | 1.7886 | 1.7589 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
112.389 |
|
C1 |
C2 |
H10 |
110.261 |
| C1 |
C2 |
H11 |
110.261 |
|
C2 |
C1 |
H7 |
111.304 |
| C2 |
C1 |
H8 |
111.283 |
|
C2 |
C1 |
H9 |
111.283 |
| C2 |
C3 |
C4 |
115.026 |
|
C2 |
C3 |
H12 |
110.735 |
| C2 |
C3 |
H13 |
110.735 |
|
C3 |
C2 |
H10 |
109.000 |
| C3 |
C2 |
H11 |
109.000 |
|
C3 |
C4 |
C5 |
116.382 |
| C3 |
C4 |
O6 |
122.197 |
|
C4 |
C3 |
H12 |
107.584 |
| C4 |
C3 |
H13 |
107.584 |
|
C4 |
C5 |
H14 |
110.204 |
| C4 |
C5 |
H15 |
110.186 |
|
C4 |
C5 |
H16 |
110.186 |
| C5 |
C4 |
O6 |
121.421 |
|
H7 |
C1 |
H8 |
107.633 |
| H7 |
C1 |
H9 |
107.633 |
|
H8 |
C1 |
H9 |
107.513 |
| H10 |
C2 |
H11 |
105.698 |
|
H12 |
C3 |
H13 |
104.591 |
| H14 |
C5 |
H15 |
109.701 |
|
H14 |
C5 |
H16 |
109.701 |
| H15 |
C5 |
H16 |
106.802 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/6-311+G(3df,2p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.478 |
|
|
|
| 2 |
C |
0.065 |
|
|
|
| 3 |
C |
-0.314 |
|
|
|
| 4 |
C |
0.486 |
|
|
|
| 5 |
C |
-0.344 |
|
|
|
| 6 |
O |
-0.714 |
|
|
|
| 7 |
H |
0.116 |
|
|
|
| 8 |
H |
0.125 |
|
|
|
| 9 |
H |
0.125 |
|
|
|
| 10 |
H |
0.118 |
|
|
|
| 11 |
H |
0.118 |
|
|
|
| 12 |
H |
0.141 |
|
|
|
| 13 |
H |
0.141 |
|
|
|
| 14 |
H |
0.128 |
|
|
|
| 15 |
H |
0.143 |
|
|
|
| 16 |
H |
0.143 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.663 |
2.675 |
0.000 |
2.756 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.837 |
1.496 |
0.000 |
| y |
1.496 |
-44.777 |
0.000 |
| z |
0.000 |
0.000 |
-37.420 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.262 |
1.496 |
0.000 |
| y |
1.496 |
-8.148 |
0.000 |
| z |
0.000 |
0.000 |
2.886 |
|
| Polar |
|---|
| 3z2-r2 | 5.773 |
|---|
| x2-y2 | 8.940 |
|---|
| xy | 1.496 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.989 |
-0.601 |
0.000 |
| y |
-0.601 |
10.446 |
0.000 |
| z |
0.000 |
0.000 |
8.118 |
<r2> (average value of r
2) Å
2
| <r2> |
229.830 |
| (<r2>)1/2 |
15.160 |