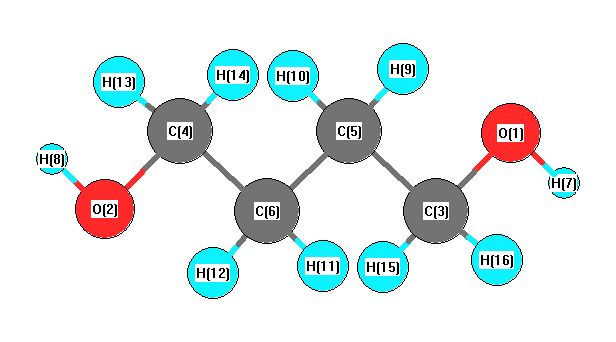
Vibrational Frequencies calculated at B97D3/6-311+G(3df,2p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3776 |
3727 |
0.00 |
|
|
|
| 2 |
Ag |
2964 |
2925 |
0.00 |
|
|
|
| 3 |
Ag |
2910 |
2872 |
0.00 |
|
|
|
| 4 |
Ag |
1490 |
1471 |
0.00 |
|
|
|
| 5 |
Ag |
1465 |
1446 |
0.00 |
|
|
|
| 6 |
Ag |
1413 |
1395 |
0.00 |
|
|
|
| 7 |
Ag |
1348 |
1330 |
0.00 |
|
|
|
| 8 |
Ag |
1237 |
1221 |
0.00 |
|
|
|
| 9 |
Ag |
1035 |
1021 |
0.00 |
|
|
|
| 10 |
Ag |
1027 |
1013 |
0.00 |
|
|
|
| 11 |
Ag |
996 |
983 |
0.00 |
|
|
|
| 12 |
Ag |
354 |
350 |
0.00 |
|
|
|
| 13 |
Ag |
331 |
327 |
0.00 |
|
|
|
| 14 |
Au |
3022 |
2982 |
88.16 |
|
|
|
| 15 |
Au |
2936 |
2898 |
78.00 |
|
|
|
| 16 |
Au |
1296 |
1279 |
1.78 |
|
|
|
| 17 |
Au |
1190 |
1175 |
1.32 |
|
|
|
| 18 |
Au |
927 |
915 |
1.05 |
|
|
|
| 19 |
Au |
740 |
730 |
0.80 |
|
|
|
| 20 |
Au |
226 |
223 |
210.68 |
|
|
|
| 21 |
Au |
78 |
77 |
19.53 |
|
|
|
| 22 |
Au |
75 |
74 |
0.34 |
|
|
|
| 23 |
Bg |
2996 |
2957 |
0.00 |
|
|
|
| 24 |
Bg |
2938 |
2899 |
0.00 |
|
|
|
| 25 |
Bg |
1291 |
1275 |
0.00 |
|
|
|
| 26 |
Bg |
1260 |
1243 |
0.00 |
|
|
|
| 27 |
Bg |
1148 |
1133 |
0.00 |
|
|
|
| 28 |
Bg |
798 |
787 |
0.00 |
|
|
|
| 29 |
Bg |
227 |
224 |
0.00 |
|
|
|
| 30 |
Bg |
143 |
141 |
0.00 |
|
|
|
| 31 |
Bu |
3776 |
3727 |
41.05 |
|
|
|
| 32 |
Bu |
2973 |
2934 |
78.02 |
|
|
|
| 33 |
Bu |
2909 |
2871 |
116.78 |
|
|
|
| 34 |
Bu |
1496 |
1477 |
5.28 |
|
|
|
| 35 |
Bu |
1472 |
1453 |
3.33 |
|
|
|
| 36 |
Bu |
1418 |
1399 |
13.50 |
|
|
|
| 37 |
Bu |
1280 |
1263 |
55.43 |
|
|
|
| 38 |
Bu |
1182 |
1167 |
36.41 |
|
|
|
| 39 |
Bu |
1022 |
1008 |
233.30 |
|
|
|
| 40 |
Bu |
957 |
945 |
3.57 |
|
|
|
| 41 |
Bu |
512 |
505 |
40.94 |
|
|
|
| 42 |
Bu |
137 |
136 |
5.58 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30384.6 cm
-1
Scaled (by 0.987) Zero Point Vibrational Energy (zpe) 29989.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/6-311+G(3df,2p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.586 |
|
|
|
| 2 |
O |
-0.586 |
|
|
|
| 3 |
C |
0.004 |
|
|
|
| 4 |
C |
0.004 |
|
|
|
| 5 |
C |
-0.092 |
|
|
|
| 6 |
C |
-0.092 |
|
|
|
| 7 |
H |
0.204 |
|
|
|
| 8 |
H |
0.204 |
|
|
|
| 9 |
H |
0.123 |
|
|
|
| 10 |
H |
0.123 |
|
|
|
| 11 |
H |
0.123 |
|
|
|
| 12 |
H |
0.123 |
|
|
|
| 13 |
H |
0.112 |
|
|
|
| 14 |
H |
0.112 |
|
|
|
| 15 |
H |
0.112 |
|
|
|
| 16 |
H |
0.112 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-27.856 |
6.004 |
0.000 |
| y |
6.004 |
-46.235 |
0.000 |
| z |
0.000 |
0.000 |
-38.914 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
14.718 |
6.004 |
0.000 |
| y |
6.004 |
-12.850 |
0.000 |
| z |
0.000 |
0.000 |
-1.869 |
|
| Polar |
|---|
| 3z2-r2 | -3.737 |
|---|
| x2-y2 | 18.379 |
|---|
| xy | 6.004 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.824 |
1.327 |
0.000 |
| y |
1.327 |
10.671 |
0.000 |
| z |
0.000 |
0.000 |
8.092 |
<r2> (average value of r
2) Å
2
| <r2> |
287.469 |
| (<r2>)1/2 |
16.955 |