Vibrational Frequencies calculated at B97D3/6-311+G(3df,2p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3040 |
3000 |
39.46 |
|
|
|
| 2 |
A' |
3037 |
2998 |
34.77 |
|
|
|
| 3 |
A' |
2990 |
2951 |
86.24 |
|
|
|
| 4 |
A' |
2967 |
2928 |
31.24 |
|
|
|
| 5 |
A' |
2960 |
2922 |
29.47 |
|
|
|
| 6 |
A' |
2945 |
2907 |
18.08 |
|
|
|
| 7 |
A' |
1502 |
1482 |
2.34 |
|
|
|
| 8 |
A' |
1475 |
1456 |
4.00 |
|
|
|
| 9 |
A' |
1460 |
1441 |
3.12 |
|
|
|
| 10 |
A' |
1394 |
1375 |
2.16 |
|
|
|
| 11 |
A' |
1372 |
1355 |
6.45 |
|
|
|
| 12 |
A' |
1340 |
1323 |
0.04 |
|
|
|
| 13 |
A' |
1263 |
1246 |
1.58 |
|
|
|
| 14 |
A' |
1233 |
1217 |
8.06 |
|
|
|
| 15 |
A' |
1133 |
1119 |
0.39 |
|
|
|
| 16 |
A' |
994 |
981 |
3.43 |
|
|
|
| 17 |
A' |
952 |
940 |
3.19 |
|
|
|
| 18 |
A' |
934 |
922 |
10.90 |
|
|
|
| 19 |
A' |
906 |
895 |
6.57 |
|
|
|
| 20 |
A' |
824 |
813 |
13.44 |
|
|
|
| 21 |
A' |
626 |
618 |
0.68 |
|
|
|
| 22 |
A' |
409 |
403 |
0.02 |
|
|
|
| 23 |
A' |
339 |
335 |
0.11 |
|
|
|
| 24 |
A' |
58 |
58 |
5.36 |
|
|
|
| 25 |
A" |
3030 |
2991 |
66.05 |
|
|
|
| 26 |
A" |
3025 |
2986 |
0.38 |
|
|
|
| 27 |
A" |
2989 |
2951 |
11.87 |
|
|
|
| 28 |
A" |
2935 |
2897 |
155.33 |
|
|
|
| 29 |
A" |
1486 |
1467 |
7.12 |
|
|
|
| 30 |
A" |
1471 |
1452 |
3.45 |
|
|
|
| 31 |
A" |
1456 |
1437 |
0.00 |
|
|
|
| 32 |
A" |
1279 |
1262 |
0.22 |
|
|
|
| 33 |
A" |
1190 |
1174 |
0.03 |
|
|
|
| 34 |
A" |
1135 |
1120 |
0.06 |
|
|
|
| 35 |
A" |
1039 |
1025 |
0.02 |
|
|
|
| 36 |
A" |
986 |
973 |
80.88 |
|
|
|
| 37 |
A" |
922 |
910 |
0.17 |
|
|
|
| 38 |
A" |
864 |
853 |
3.34 |
|
|
|
| 39 |
A" |
385 |
380 |
0.89 |
|
|
|
| 40 |
A" |
309 |
305 |
0.11 |
|
|
|
| 41 |
A" |
248 |
245 |
0.08 |
|
|
|
| 42 |
A" |
206 |
204 |
0.01 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30553.6 cm
-1
Scaled (by 0.987) Zero Point Vibrational Energy (zpe) 30156.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
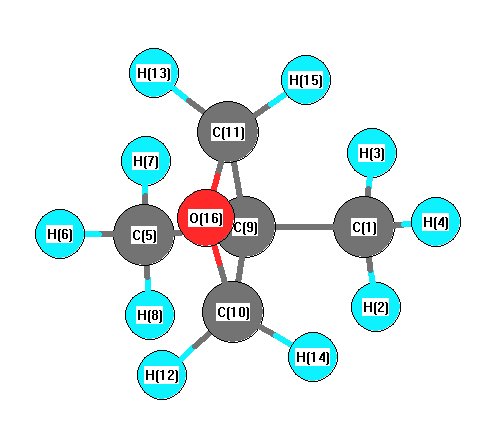
Charges from optimized geometry at B97D3/6-311+G(3df,2p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.302 |
|
|
|
| 2 |
H |
0.121 |
|
|
|
| 3 |
H |
0.121 |
|
|
|
| 4 |
H |
0.124 |
|
|
|
| 5 |
C |
-0.339 |
|
|
|
| 6 |
H |
0.128 |
|
|
|
| 7 |
H |
0.123 |
|
|
|
| 8 |
H |
0.123 |
|
|
|
| 9 |
C |
-0.285 |
|
|
|
| 10 |
C |
0.275 |
|
|
|
| 11 |
C |
0.275 |
|
|
|
| 12 |
H |
0.093 |
|
|
|
| 13 |
H |
0.093 |
|
|
|
| 14 |
H |
0.090 |
|
|
|
| 15 |
H |
0.090 |
|
|
|
| 16 |
O |
-0.729 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.250 |
1.989 |
0.000 |
2.004 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-38.349 |
0.590 |
0.000 |
| y |
0.590 |
-44.115 |
0.000 |
| z |
0.000 |
0.000 |
-36.611 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.014 |
0.590 |
0.000 |
| y |
0.590 |
-6.635 |
0.000 |
| z |
0.000 |
0.000 |
4.621 |
|
| Polar |
|---|
| 3z2-r2 | 9.242 |
|---|
| x2-y2 | 5.766 |
|---|
| xy | 0.590 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.747 |
-0.019 |
0.000 |
| y |
-0.019 |
10.066 |
0.000 |
| z |
0.000 |
0.000 |
9.853 |
<r2> (average value of r
2) Å
2
| <r2> |
159.802 |
| (<r2>)1/2 |
12.641 |