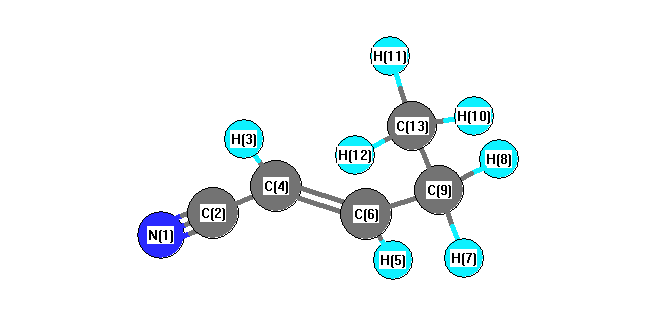
Vibrational Frequencies calculated at B97D3/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3123 |
3075 |
2.81 |
|
|
|
| 2 |
A' |
3100 |
3052 |
7.63 |
|
|
|
| 3 |
A' |
3059 |
3013 |
27.27 |
|
|
|
| 4 |
A' |
2986 |
2940 |
20.93 |
|
|
|
| 5 |
A' |
2951 |
2906 |
18.17 |
|
|
|
| 6 |
A' |
2245 |
2211 |
28.02 |
|
|
|
| 7 |
A' |
1646 |
1620 |
29.56 |
|
|
|
| 8 |
A' |
1480 |
1457 |
4.65 |
|
|
|
| 9 |
A' |
1430 |
1408 |
16.60 |
|
|
|
| 10 |
A' |
1391 |
1370 |
3.95 |
|
|
|
| 11 |
A' |
1352 |
1332 |
7.40 |
|
|
|
| 12 |
A' |
1310 |
1290 |
2.19 |
|
|
|
| 13 |
A' |
1271 |
1251 |
1.19 |
|
|
|
| 14 |
A' |
1089 |
1073 |
1.87 |
|
|
|
| 15 |
A' |
1049 |
1033 |
6.62 |
|
|
|
| 16 |
A' |
972 |
957 |
0.91 |
|
|
|
| 17 |
A' |
877 |
864 |
5.10 |
|
|
|
| 18 |
A' |
569 |
560 |
0.62 |
|
|
|
| 19 |
A' |
501 |
494 |
0.01 |
|
|
|
| 20 |
A' |
257 |
253 |
1.18 |
|
|
|
| 21 |
A' |
141 |
139 |
4.12 |
|
|
|
| 22 |
A" |
3050 |
3004 |
27.32 |
|
|
|
| 23 |
A" |
2967 |
2922 |
8.78 |
|
|
|
| 24 |
A" |
1476 |
1453 |
7.48 |
|
|
|
| 25 |
A" |
1263 |
1244 |
0.27 |
|
|
|
| 26 |
A" |
1079 |
1062 |
1.47 |
|
|
|
| 27 |
A" |
969 |
954 |
30.98 |
|
|
|
| 28 |
A" |
831 |
818 |
6.69 |
|
|
|
| 29 |
A" |
719 |
708 |
0.41 |
|
|
|
| 30 |
A" |
473 |
466 |
4.44 |
|
|
|
| 31 |
A" |
273 |
268 |
0.01 |
|
|
|
| 32 |
A" |
173 |
170 |
0.06 |
|
|
|
| 33 |
A" |
118 |
117 |
3.68 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23095.1 cm
-1
Scaled (by 0.9847) Zero Point Vibrational Energy (zpe) 22741.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.789 |
|
|
|
| 2 |
C |
0.709 |
|
|
|
| 3 |
H |
0.101 |
|
|
|
| 4 |
C |
0.182 |
|
|
|
| 5 |
H |
0.020 |
|
|
|
| 6 |
C |
-0.509 |
|
|
|
| 7 |
H |
0.126 |
|
|
|
| 8 |
H |
0.126 |
|
|
|
| 9 |
C |
0.101 |
|
|
|
| 10 |
H |
0.018 |
|
|
|
| 11 |
H |
0.164 |
|
|
|
| 12 |
H |
0.164 |
|
|
|
| 13 |
C |
-0.415 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.203 |
-4.546 |
0.000 |
5.051 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-38.465 |
7.059 |
0.000 |
| y |
7.059 |
-46.034 |
0.000 |
| z |
0.000 |
0.000 |
-37.407 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.255 |
7.059 |
0.000 |
| y |
7.059 |
-8.098 |
0.000 |
| z |
0.000 |
0.000 |
4.843 |
|
| Polar |
|---|
| 3z2-r2 | 9.686 |
|---|
| x2-y2 | 7.568 |
|---|
| xy | 7.059 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.849 |
-2.145 |
0.000 |
| y |
-2.145 |
14.703 |
0.000 |
| z |
0.000 |
0.000 |
7.333 |
<r2> (average value of r
2) Å
2
| <r2> |
227.327 |
| (<r2>)1/2 |
15.077 |