Jump to
S1C2
Energy calculated at B97D3/aug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -189.218668 |
| Energy at 298.15K | -189.225942 |
| HF Energy | -189.218668 |
| Nuclear repulsion energy | 121.839102 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B97D3/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3093 |
3045 |
0.79 |
|
|
|
| 2 |
A1 |
2960 |
2915 |
18.41 |
|
|
|
| 3 |
A1 |
1585 |
1561 |
14.74 |
|
|
|
| 4 |
A1 |
1440 |
1418 |
0.05 |
|
|
|
| 5 |
A1 |
1355 |
1334 |
20.40 |
|
|
|
| 6 |
A1 |
1065 |
1049 |
4.46 |
|
|
|
| 7 |
A1 |
812 |
800 |
0.30 |
|
|
|
| 8 |
A1 |
353 |
347 |
1.27 |
|
|
|
| 9 |
A2 |
3029 |
2983 |
0.00 |
|
|
|
| 10 |
A2 |
1449 |
1427 |
0.00 |
|
|
|
| 11 |
A2 |
1058 |
1042 |
0.00 |
|
|
|
| 12 |
A2 |
470 |
462 |
0.00 |
|
|
|
| 13 |
A2 |
78 |
77 |
0.00 |
|
|
|
| 14 |
B1 |
3021 |
2975 |
34.66 |
|
|
|
| 15 |
B1 |
1471 |
1449 |
20.95 |
|
|
|
| 16 |
B1 |
905 |
891 |
3.69 |
|
|
|
| 17 |
B1 |
171 |
169 |
0.15 |
|
|
|
| 18 |
B2 |
3092 |
3044 |
35.06 |
|
|
|
| 19 |
B2 |
2960 |
2915 |
3.13 |
|
|
|
| 20 |
B2 |
1430 |
1409 |
10.51 |
|
|
|
| 21 |
B2 |
1344 |
1323 |
1.13 |
|
|
|
| 22 |
B2 |
1146 |
1129 |
15.12 |
|
|
|
| 23 |
B2 |
882 |
869 |
21.07 |
|
|
|
| 24 |
B2 |
611 |
602 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 17890.0 cm
-1
Scaled (by 0.9847) Zero Point Vibrational Energy (zpe) 17616.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B97D3/aug-cc-pVTZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.620 |
-0.783 |
| N2 |
0.000 |
-0.620 |
-0.783 |
| C3 |
0.000 |
1.366 |
0.503 |
| C4 |
0.000 |
-1.366 |
0.503 |
| H5 |
0.000 |
2.430 |
0.265 |
| H6 |
0.000 |
-2.430 |
0.265 |
| H7 |
-0.888 |
1.125 |
1.100 |
| H8 |
0.888 |
1.125 |
1.100 |
| H9 |
0.888 |
-1.125 |
1.100 |
| H10 |
-0.888 |
-1.125 |
1.100 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| N1 | | 1.2393 | 1.4867 | 2.3657 | 2.0918 | 3.2247 | 2.1426 | 2.1426 | 2.7165 | 2.7165 |
N2 | 1.2393 | | 2.3657 | 1.4867 | 3.2247 | 2.0918 | 2.7165 | 2.7165 | 2.1426 | 2.1426 | C3 | 1.4867 | 2.3657 | | 2.7324 | 1.0899 | 3.8035 | 1.0973 | 1.0973 | 2.7115 | 2.7115 | C4 | 2.3657 | 1.4867 | 2.7324 | | 3.8035 | 1.0899 | 2.7115 | 2.7115 | 1.0973 | 1.0973 | H5 | 2.0918 | 3.2247 | 1.0899 | 3.8035 | | 4.8598 | 1.7857 | 1.7857 | 3.7582 | 3.7582 | H6 | 3.2247 | 2.0918 | 3.8035 | 1.0899 | 4.8598 | | 3.7582 | 3.7582 | 1.7857 | 1.7857 | H7 | 2.1426 | 2.7165 | 1.0973 | 2.7115 | 1.7857 | 3.7582 | | 1.7764 | 2.8669 | 2.2502 | H8 | 2.1426 | 2.7165 | 1.0973 | 2.7115 | 1.7857 | 3.7582 | 1.7764 | | 2.2502 | 2.8669 | H9 | 2.7165 | 2.1426 | 2.7115 | 1.0973 | 3.7582 | 1.7857 | 2.8669 | 2.2502 | | 1.7764 | H10 | 2.7165 | 2.1426 | 2.7115 | 1.0973 | 3.7582 | 1.7857 | 2.2502 | 2.8669 | 1.7764 | |
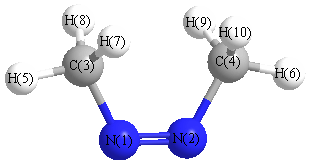 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
N2 |
C4 |
120.143 |
|
N1 |
C3 |
H5 |
107.560 |
| N1 |
C3 |
H7 |
111.134 |
|
N1 |
C3 |
H8 |
111.134 |
| N2 |
N1 |
C3 |
120.143 |
|
N2 |
C4 |
H6 |
107.560 |
| N2 |
C4 |
H9 |
111.134 |
|
N2 |
C4 |
H10 |
111.134 |
| H5 |
C3 |
H7 |
109.457 |
|
H5 |
C3 |
H8 |
109.457 |
| H6 |
C4 |
H9 |
109.457 |
|
H6 |
C4 |
H10 |
109.457 |
| H7 |
C3 |
H8 |
108.084 |
|
H9 |
C4 |
H10 |
108.084 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.204 |
|
|
|
| 2 |
N |
-0.204 |
|
|
|
| 3 |
C |
-0.302 |
|
|
|
| 4 |
C |
-0.302 |
|
|
|
| 5 |
H |
0.124 |
|
|
|
| 6 |
H |
0.124 |
|
|
|
| 7 |
H |
0.191 |
|
|
|
| 8 |
H |
0.191 |
|
|
|
| 9 |
H |
0.191 |
|
|
|
| 10 |
H |
0.191 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
3.098 |
3.098 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.771 |
0.000 |
0.000 |
| y |
0.000 |
-23.883 |
0.000 |
| z |
0.000 |
0.000 |
-29.378 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.860 |
0.000 |
0.000 |
| y |
0.000 |
3.191 |
0.000 |
| z |
0.000 |
0.000 |
-5.050 |
|
| Polar |
|---|
| 3z2-r2 | -10.101 |
|---|
| x2-y2 | -0.887 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.346 |
0.000 |
0.000 |
| y |
0.000 |
8.771 |
0.000 |
| z |
0.000 |
0.000 |
6.807 |
<r2> (average value of r
2) Å
2
| <r2> |
80.645 |
| (<r2>)1/2 |
8.980 |
Jump to
S1C1
Energy calculated at B97D3/aug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -189.218668 |
| Energy at 298.15K | -189.225939 |
| HF Energy | -189.218668 |
| Nuclear repulsion energy | 121.827231 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B97D3/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3093 |
3046 |
0.79 |
|
|
|
| 2 |
A |
3030 |
2983 |
0.00 |
|
|
|
| 3 |
A |
2961 |
2915 |
18.36 |
|
|
|
| 4 |
A |
1585 |
1561 |
14.82 |
|
|
|
| 5 |
A |
1449 |
1427 |
0.00 |
|
|
|
| 6 |
A |
1440 |
1418 |
0.05 |
|
|
|
| 7 |
A |
1355 |
1334 |
20.41 |
|
|
|
| 8 |
A |
1065 |
1049 |
4.45 |
|
|
|
| 9 |
A |
1058 |
1041 |
0.00 |
|
|
|
| 10 |
A |
811 |
798 |
0.32 |
|
|
|
| 11 |
A |
469 |
462 |
0.00 |
|
|
|
| 12 |
A |
353 |
347 |
1.27 |
|
|
|
| 13 |
A |
78 |
77 |
0.00 |
|
|
|
| 14 |
B |
3092 |
3045 |
34.93 |
|
|
|
| 15 |
B |
3022 |
2976 |
34.62 |
|
|
|
| 16 |
B |
2961 |
2916 |
3.12 |
|
|
|
| 17 |
B |
1471 |
1449 |
20.96 |
|
|
|
| 18 |
B |
1430 |
1408 |
10.54 |
|
|
|
| 19 |
B |
1343 |
1323 |
1.11 |
|
|
|
| 20 |
B |
1146 |
1128 |
15.18 |
|
|
|
| 21 |
B |
905 |
891 |
3.70 |
|
|
|
| 22 |
B |
881 |
868 |
21.16 |
|
|
|
| 23 |
B |
611 |
601 |
0.00 |
|
|
|
| 24 |
B |
171 |
169 |
0.15 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 17888.9 cm
-1
Scaled (by 0.9847) Zero Point Vibrational Energy (zpe) 17615.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B97D3/aug-cc-pVTZ
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.620 |
-0.783 |
| N2 |
0.000 |
-0.620 |
-0.783 |
| C3 |
0.000 |
1.366 |
0.503 |
| C4 |
0.000 |
-1.366 |
0.503 |
| H5 |
-0.001 |
2.430 |
0.265 |
| H6 |
0.001 |
-2.430 |
0.265 |
| H7 |
-0.888 |
1.124 |
1.101 |
| H8 |
0.889 |
1.125 |
1.100 |
| H9 |
0.888 |
-1.124 |
1.101 |
| H10 |
-0.889 |
-1.125 |
1.100 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| N1 | | 1.2391 | 1.4872 | 2.3660 | 2.0919 | 3.2246 | 2.1429 | 2.1428 | 2.7163 | 2.7167 |
N2 | 1.2391 | | 2.3660 | 1.4872 | 3.2246 | 2.0919 | 2.7163 | 2.7167 | 2.1429 | 2.1428 | C3 | 1.4872 | 2.3660 | | 2.7328 | 1.0898 | 3.8038 | 1.0973 | 1.0973 | 2.7110 | 2.7121 | C4 | 2.3660 | 1.4872 | 2.7328 | | 3.8038 | 1.0898 | 2.7110 | 2.7121 | 1.0973 | 1.0973 | H5 | 2.0919 | 3.2246 | 1.0898 | 3.8038 | | 4.8599 | 1.7859 | 1.7859 | 3.7578 | 3.7585 | H6 | 3.2246 | 2.0919 | 3.8038 | 1.0898 | 4.8599 | | 3.7578 | 3.7585 | 1.7859 | 1.7859 | H7 | 2.1429 | 2.7163 | 1.0973 | 2.7110 | 1.7859 | 3.7578 | | 1.7764 | 2.8653 | 2.2499 | H8 | 2.1428 | 2.7167 | 1.0973 | 2.7121 | 1.7859 | 3.7585 | 1.7764 | | 2.2499 | 2.8680 | H9 | 2.7163 | 2.1429 | 2.7110 | 1.0973 | 3.7578 | 1.7859 | 2.8653 | 2.2499 | | 1.7764 | H10 | 2.7167 | 2.1428 | 2.7121 | 1.0973 | 3.7585 | 1.7859 | 2.2499 | 2.8680 | 1.7764 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
N2 |
C4 |
120.143 |
|
N1 |
C3 |
H5 |
107.535 |
| N1 |
C3 |
H7 |
111.120 |
|
N1 |
C3 |
H8 |
111.111 |
| N2 |
N1 |
C3 |
120.143 |
|
N2 |
C4 |
H6 |
107.535 |
| N2 |
C4 |
H9 |
111.120 |
|
N2 |
C4 |
H10 |
111.111 |
| H5 |
C3 |
H7 |
109.489 |
|
H5 |
C3 |
H8 |
109.483 |
| H6 |
C4 |
H9 |
109.489 |
|
H6 |
C4 |
H10 |
109.483 |
| H7 |
C3 |
H8 |
108.088 |
|
H9 |
C4 |
H10 |
108.088 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.204 |
|
|
|
| 2 |
N |
-0.204 |
|
|
|
| 3 |
C |
-0.303 |
|
|
|
| 4 |
C |
-0.303 |
|
|
|
| 5 |
H |
0.124 |
|
|
|
| 6 |
H |
0.124 |
|
|
|
| 7 |
H |
0.191 |
|
|
|
| 8 |
H |
0.191 |
|
|
|
| 9 |
H |
0.191 |
|
|
|
| 10 |
H |
0.191 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
3.097 |
3.097 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.771 |
0.000 |
0.000 |
| y |
0.000 |
-23.885 |
0.000 |
| z |
0.000 |
0.000 |
-29.378 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.860 |
0.000 |
0.000 |
| y |
0.000 |
3.190 |
0.000 |
| z |
0.000 |
0.000 |
-5.050 |
|
| Polar |
|---|
| 3z2-r2 | -10.100 |
|---|
| x2-y2 | -0.886 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.346 |
0.000 |
0.000 |
| y |
0.000 |
8.773 |
0.000 |
| z |
0.000 |
0.000 |
6.808 |
<r2> (average value of r
2) Å
2
| <r2> |
80.657 |
| (<r2>)1/2 |
8.981 |