Jump to
S1C2
Energy calculated at B97D3/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -339.575921 |
| Energy at 298.15K | -339.584790 |
| HF Energy | -339.575921 |
| Nuclear repulsion energy | 258.172962 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B97D3/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3150 |
3106 |
0.00 |
|
|
|
| 2 |
Ag |
3013 |
2971 |
0.00 |
|
|
|
| 3 |
Ag |
1507 |
1486 |
0.00 |
|
|
|
| 4 |
Ag |
1422 |
1402 |
0.00 |
|
|
|
| 5 |
Ag |
1393 |
1373 |
0.00 |
|
|
|
| 6 |
Ag |
1311 |
1293 |
0.00 |
|
|
|
| 7 |
Ag |
1078 |
1063 |
0.00 |
|
|
|
| 8 |
Ag |
750 |
740 |
0.00 |
|
|
|
| 9 |
Ag |
600 |
591 |
0.00 |
|
|
|
| 10 |
Ag |
401 |
396 |
0.00 |
|
|
|
| 11 |
Au |
3082 |
3039 |
15.32 |
|
|
|
| 12 |
Au |
1437 |
1417 |
21.43 |
|
|
|
| 13 |
Au |
1100 |
1085 |
0.00 |
|
|
|
| 14 |
Au |
323 |
318 |
4.87 |
|
|
|
| 15 |
Au |
175 |
173 |
10.43 |
|
|
|
| 16 |
Au |
110 |
109 |
3.51 |
|
|
|
| 17 |
Bg |
3082 |
3039 |
0.00 |
|
|
|
| 18 |
Bg |
1434 |
1414 |
0.00 |
|
|
|
| 19 |
Bg |
1073 |
1058 |
0.00 |
|
|
|
| 20 |
Bg |
462 |
456 |
0.00 |
|
|
|
| 21 |
Bg |
146 |
144 |
0.00 |
|
|
|
| 22 |
Bu |
3151 |
3106 |
1.73 |
|
|
|
| 23 |
Bu |
3013 |
2971 |
15.87 |
|
|
|
| 24 |
Bu |
1452 |
1432 |
0.70 |
|
|
|
| 25 |
Bu |
1399 |
1380 |
34.55 |
|
|
|
| 26 |
Bu |
1321 |
1303 |
286.43 |
|
|
|
| 27 |
Bu |
1122 |
1106 |
49.30 |
|
|
|
| 28 |
Bu |
916 |
904 |
23.51 |
|
|
|
| 29 |
Bu |
538 |
530 |
33.01 |
|
|
|
| 30 |
Bu |
291 |
287 |
22.10 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20126.6 cm
-1
Scaled (by 0.986) Zero Point Vibrational Energy (zpe) 19844.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B97D3/cc-pVTZ
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
-0.066 |
0.657 |
0.000 |
| N2 |
0.066 |
-0.657 |
0.000 |
| O3 |
-1.198 |
1.218 |
0.000 |
| O4 |
1.198 |
-1.218 |
0.000 |
| C5 |
1.198 |
1.421 |
0.000 |
| C6 |
-1.198 |
-1.421 |
0.000 |
| H7 |
0.904 |
2.467 |
0.000 |
| H8 |
1.781 |
1.158 |
0.886 |
| H9 |
1.781 |
1.158 |
-0.886 |
| H10 |
-0.904 |
-2.467 |
0.000 |
| H11 |
-1.781 |
-1.158 |
0.886 |
| H12 |
-1.781 |
-1.158 |
-0.886 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
| N1 | | 1.3217 | 1.2625 | 2.2617 | 1.4766 | 2.3661 | 2.0532 | 2.1089 | 2.1089 | 3.2347 | 2.6495 | 2.6495 |
N2 | 1.3217 | | 2.2617 | 1.2625 | 2.3661 | 1.4766 | 3.2347 | 2.6495 | 2.6495 | 2.0532 | 2.1089 | 2.1089 | O3 | 1.2625 | 2.2617 | | 3.4163 | 2.4039 | 2.6386 | 2.4448 | 3.1080 | 3.1080 | 3.6965 | 2.6019 | 2.6019 | O4 | 2.2617 | 1.2625 | 3.4163 | | 2.6386 | 2.4039 | 3.6965 | 2.6019 | 2.6019 | 2.4448 | 3.1080 | 3.1080 | C5 | 1.4766 | 2.3661 | 2.4039 | 2.6386 | | 3.7162 | 1.0867 | 1.0924 | 1.0924 | 4.4193 | 4.0380 | 4.0380 | C6 | 2.3661 | 1.4766 | 2.6386 | 2.4039 | 3.7162 | | 4.4193 | 4.0380 | 4.0380 | 1.0867 | 1.0924 | 1.0924 | H7 | 2.0532 | 3.2347 | 2.4448 | 3.6965 | 1.0867 | 4.4193 | | 1.8073 | 1.8073 | 5.2547 | 4.5971 | 4.5971 | H8 | 2.1089 | 2.6495 | 3.1080 | 2.6019 | 1.0924 | 4.0380 | 1.8073 | | 1.7713 | 4.5971 | 4.2485 | 4.6030 | H9 | 2.1089 | 2.6495 | 3.1080 | 2.6019 | 1.0924 | 4.0380 | 1.8073 | 1.7713 | | 4.5971 | 4.6030 | 4.2485 | H10 | 3.2347 | 2.0532 | 3.6965 | 2.4448 | 4.4193 | 1.0867 | 5.2547 | 4.5971 | 4.5971 | | 1.8073 | 1.8073 | H11 | 2.6495 | 2.1089 | 2.6019 | 3.1080 | 4.0380 | 1.0924 | 4.5971 | 4.2485 | 4.6030 | 1.8073 | | 1.7713 | H12 | 2.6495 | 2.1089 | 2.6019 | 3.1080 | 4.0380 | 1.0924 | 4.5971 | 4.6030 | 4.2485 | 1.8073 | 1.7713 | |
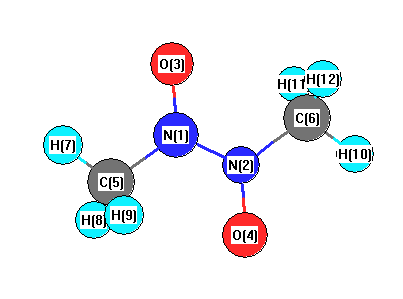 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
N2 |
O4 |
123.458 |
|
N1 |
N2 |
C6 |
115.135 |
| N1 |
C5 |
H7 |
105.104 |
|
N1 |
C5 |
H8 |
109.986 |
| N1 |
C5 |
H9 |
109.986 |
|
N2 |
N1 |
O3 |
123.458 |
| N2 |
N1 |
C5 |
115.135 |
|
N2 |
C6 |
H10 |
105.104 |
| N2 |
C6 |
H11 |
109.986 |
|
N2 |
C6 |
H12 |
109.986 |
| O3 |
N1 |
C5 |
121.408 |
|
O4 |
N2 |
C6 |
121.408 |
| H7 |
C5 |
H8 |
111.465 |
|
H7 |
C5 |
H9 |
111.465 |
| H8 |
C5 |
H9 |
108.795 |
|
H10 |
C6 |
H11 |
111.465 |
| H10 |
C6 |
H12 |
111.465 |
|
H11 |
C6 |
H12 |
108.795 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
0.195 |
|
|
|
| 2 |
N |
0.195 |
|
|
|
| 3 |
O |
-0.379 |
|
|
|
| 4 |
O |
-0.379 |
|
|
|
| 5 |
C |
-0.136 |
|
|
|
| 6 |
C |
-0.136 |
|
|
|
| 7 |
H |
0.106 |
|
|
|
| 8 |
H |
0.107 |
|
|
|
| 9 |
H |
0.107 |
|
|
|
| 10 |
H |
0.106 |
|
|
|
| 11 |
H |
0.107 |
|
|
|
| 12 |
H |
0.107 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-37.299 |
9.563 |
0.000 |
| y |
9.563 |
-35.198 |
0.000 |
| z |
0.000 |
0.000 |
-34.282 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.559 |
9.563 |
0.000 |
| y |
9.563 |
0.593 |
0.000 |
| z |
0.000 |
0.000 |
1.967 |
|
| Polar |
|---|
| 3z2-r2 | 3.933 |
|---|
| x2-y2 | -2.101 |
|---|
| xy | 9.563 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.756 |
-0.312 |
0.000 |
| y |
-0.312 |
9.957 |
0.000 |
| z |
0.000 |
0.000 |
5.175 |
<r2> (average value of r
2) Å
2
| <r2> |
151.454 |
| (<r2>)1/2 |
12.307 |
Jump to
S1C1
Energy calculated at B97D3/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -339.575914 |
| Energy at 298.15K | -339.584766 |
| HF Energy | -339.575914 |
| Nuclear repulsion energy | 258.157856 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B97D3/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3148 |
3104 |
0.00 |
|
|
|
| 2 |
Ag |
3082 |
3039 |
0.00 |
|
|
|
| 3 |
Ag |
3013 |
2970 |
0.00 |
|
|
|
| 4 |
Ag |
1507 |
1486 |
0.00 |
|
|
|
| 5 |
Ag |
1434 |
1414 |
0.00 |
|
|
|
| 6 |
Ag |
1421 |
1402 |
0.00 |
|
|
|
| 7 |
Ag |
1391 |
1372 |
0.00 |
|
|
|
| 8 |
Ag |
1310 |
1291 |
0.00 |
|
|
|
| 9 |
Ag |
1077 |
1062 |
0.00 |
|
|
|
| 10 |
Ag |
1072 |
1057 |
0.00 |
|
|
|
| 11 |
Ag |
750 |
739 |
0.00 |
|
|
|
| 12 |
Ag |
599 |
591 |
0.00 |
|
|
|
| 13 |
Ag |
461 |
454 |
0.00 |
|
|
|
| 14 |
Ag |
400 |
394 |
0.00 |
|
|
|
| 15 |
Ag |
146 |
144 |
0.00 |
|
|
|
| 16 |
Au |
3149 |
3105 |
1.71 |
|
|
|
| 17 |
Au |
3082 |
3039 |
15.30 |
|
|
|
| 18 |
Au |
3013 |
2971 |
15.94 |
|
|
|
| 19 |
Au |
1452 |
1432 |
0.76 |
|
|
|
| 20 |
Au |
1437 |
1417 |
21.43 |
|
|
|
| 21 |
Au |
1397 |
1378 |
37.31 |
|
|
|
| 22 |
Au |
1321 |
1303 |
283.92 |
|
|
|
| 23 |
Au |
1121 |
1105 |
49.16 |
|
|
|
| 24 |
Au |
1099 |
1084 |
0.00 |
|
|
|
| 25 |
Au |
916 |
903 |
23.70 |
|
|
|
| 26 |
Au |
538 |
530 |
33.01 |
|
|
|
| 27 |
Au |
321 |
317 |
5.08 |
|
|
|
| 28 |
Au |
289 |
285 |
21.97 |
|
|
|
| 29 |
Au |
174 |
172 |
9.88 |
|
|
|
| 30 |
Au |
108 |
106 |
3.83 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20112.6 cm
-1
Scaled (by 0.986) Zero Point Vibrational Energy (zpe) 19831.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B97D3/cc-pVTZ
Point Group is Ci
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
-0.554 |
0.257 |
-0.252 |
| N2 |
0.554 |
-0.257 |
0.252 |
| O3 |
-0.573 |
1.380 |
-0.828 |
| O4 |
0.573 |
-1.380 |
0.828 |
| C5 |
-1.765 |
-0.574 |
-0.094 |
| C6 |
1.765 |
0.574 |
0.094 |
| H7 |
-2.568 |
-0.006 |
-0.557 |
| H8 |
-1.610 |
-1.538 |
-0.584 |
| H9 |
-1.948 |
-0.752 |
0.968 |
| H10 |
1.948 |
0.752 |
-0.968 |
| H11 |
1.610 |
1.538 |
0.584 |
| H12 |
2.568 |
0.006 |
0.557 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
| N1 | | 1.3221 | 1.2624 | 2.2622 | 1.4766 | 2.3662 | 2.0532 | 2.1090 | 2.1089 | 2.6492 | 2.6505 | 3.2350 |
N2 | 1.3221 | | 2.2622 | 1.2624 | 2.3662 | 1.4766 | 3.2350 | 2.6505 | 2.6492 | 2.1089 | 2.1090 | 2.0532 | O3 | 1.2624 | 2.2622 | | 3.4168 | 2.4037 | 2.6390 | 2.4445 | 3.1070 | 3.1084 | 2.6014 | 2.6044 | 3.6971 | O4 | 2.2622 | 1.2624 | 3.4168 | | 2.6390 | 2.4037 | 3.6971 | 2.6044 | 2.6014 | 3.1084 | 3.1070 | 2.4445 | C5 | 1.4766 | 2.3662 | 2.4037 | 2.6390 | | 3.7162 | 1.0868 | 1.0924 | 1.0923 | 4.0378 | 4.0387 | 4.4193 | C6 | 2.3662 | 1.4766 | 2.6390 | 2.4037 | 3.7162 | | 4.4193 | 4.0387 | 4.0378 | 1.0923 | 1.0924 | 1.0868 | H7 | 2.0532 | 3.2350 | 2.4445 | 3.6971 | 1.0868 | 4.4193 | | 1.8067 | 1.8072 | 4.5970 | 4.5980 | 5.2548 | H8 | 2.1090 | 2.6505 | 3.1070 | 2.6044 | 1.0924 | 4.0387 | 1.8067 | | 1.7718 | 4.2488 | 4.6041 | 4.5980 | H9 | 2.1089 | 2.6492 | 3.1084 | 2.6014 | 1.0923 | 4.0378 | 1.8072 | 1.7718 | | 4.6026 | 4.2488 | 4.5970 | H10 | 2.6492 | 2.1089 | 2.6014 | 3.1084 | 4.0378 | 1.0923 | 4.5970 | 4.2488 | 4.6026 | | 1.7718 | 1.8072 | H11 | 2.6505 | 2.1090 | 2.6044 | 3.1070 | 4.0387 | 1.0924 | 4.5980 | 4.6041 | 4.2488 | 1.7718 | | 1.8067 | H12 | 3.2350 | 2.0532 | 3.6971 | 2.4445 | 4.4193 | 1.0868 | 5.2548 | 4.5980 | 4.5970 | 1.8072 | 1.8067 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
N2 |
O4 |
119.170 |
|
N1 |
N2 |
C6 |
112.213 |
| N1 |
C5 |
H7 |
109.215 |
|
N1 |
C5 |
H8 |
104.109 |
| N1 |
C5 |
H9 |
108.224 |
|
N2 |
N1 |
O3 |
119.170 |
| N2 |
N1 |
C5 |
112.213 |
|
N2 |
C6 |
H10 |
108.224 |
| N2 |
C6 |
H11 |
104.109 |
|
N2 |
C6 |
H12 |
109.215 |
| O3 |
N1 |
C5 |
122.528 |
|
O4 |
N2 |
C6 |
122.528 |
| H7 |
C5 |
H8 |
112.920 |
|
H7 |
C5 |
H9 |
110.695 |
| H8 |
C5 |
H9 |
111.360 |
|
H10 |
C6 |
H11 |
111.360 |
| H10 |
C6 |
H12 |
110.695 |
|
H11 |
C6 |
H12 |
112.920 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
0.195 |
|
|
|
| 2 |
N |
0.195 |
|
|
|
| 3 |
O |
-0.379 |
|
|
|
| 4 |
O |
-0.379 |
|
|
|
| 5 |
C |
-0.136 |
|
|
|
| 6 |
C |
-0.136 |
|
|
|
| 7 |
H |
0.105 |
|
|
|
| 8 |
H |
0.107 |
|
|
|
| 9 |
H |
0.107 |
|
|
|
| 10 |
H |
0.107 |
|
|
|
| 11 |
H |
0.107 |
|
|
|
| 12 |
H |
0.105 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-28.379 |
5.068 |
-1.284 |
| y |
5.068 |
-41.427 |
4.744 |
| z |
-1.284 |
4.744 |
-36.973 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
10.821 |
5.068 |
-1.284 |
| y |
5.068 |
-8.751 |
4.744 |
| z |
-1.284 |
4.744 |
-2.070 |
|
| Polar |
|---|
| 3z2-r2 | -4.140 |
|---|
| x2-y2 | 13.048 |
|---|
| xy | 5.068 |
|---|
| xz | -1.284 |
|---|
| yz | 4.744 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.334 |
-0.214 |
1.021 |
| y |
-0.214 |
8.320 |
-1.646 |
| z |
1.021 |
-1.646 |
6.236 |
<r2> (average value of r
2) Å
2
| <r2> |
151.475 |
| (<r2>)1/2 |
12.308 |