Vibrational Frequencies calculated at B97D3/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3509 |
3460 |
0.76 |
|
|
|
| 2 |
A |
3508 |
3459 |
0.51 |
|
|
|
| 3 |
A |
3425 |
3377 |
3.09 |
|
|
|
| 4 |
A |
3420 |
3372 |
6.14 |
|
|
|
| 5 |
A |
3049 |
3007 |
50.03 |
|
|
|
| 6 |
A |
3040 |
2998 |
45.72 |
|
|
|
| 7 |
A |
3020 |
2978 |
14.46 |
|
|
|
| 8 |
A |
2990 |
2948 |
59.42 |
|
|
|
| 9 |
A |
2984 |
2942 |
7.55 |
|
|
|
| 10 |
A |
2968 |
2926 |
35.80 |
|
|
|
| 11 |
A |
2868 |
2828 |
133.21 |
|
|
|
| 12 |
A |
2848 |
2809 |
75.99 |
|
|
|
| 13 |
A |
1625 |
1602 |
31.60 |
|
|
|
| 14 |
A |
1608 |
1585 |
37.82 |
|
|
|
| 15 |
A |
1482 |
1461 |
5.49 |
|
|
|
| 16 |
A |
1481 |
1460 |
6.17 |
|
|
|
| 17 |
A |
1471 |
1450 |
3.69 |
|
|
|
| 18 |
A |
1454 |
1434 |
1.50 |
|
|
|
| 19 |
A |
1396 |
1376 |
9.46 |
|
|
|
| 20 |
A |
1392 |
1373 |
2.54 |
|
|
|
| 21 |
A |
1381 |
1362 |
0.90 |
|
|
|
| 22 |
A |
1353 |
1334 |
11.28 |
|
|
|
| 23 |
A |
1319 |
1301 |
2.04 |
|
|
|
| 24 |
A |
1280 |
1262 |
0.53 |
|
|
|
| 25 |
A |
1260 |
1242 |
1.86 |
|
|
|
| 26 |
A |
1240 |
1223 |
1.61 |
|
|
|
| 27 |
A |
1168 |
1152 |
3.62 |
|
|
|
| 28 |
A |
1123 |
1107 |
4.63 |
|
|
|
| 29 |
A |
1077 |
1062 |
1.40 |
|
|
|
| 30 |
A |
1051 |
1037 |
5.32 |
|
|
|
| 31 |
A |
1032 |
1017 |
2.37 |
|
|
|
| 32 |
A |
989 |
975 |
1.20 |
|
|
|
| 33 |
A |
949 |
936 |
20.18 |
|
|
|
| 34 |
A |
900 |
887 |
19.18 |
|
|
|
| 35 |
A |
889 |
877 |
55.33 |
|
|
|
| 36 |
A |
833 |
821 |
123.49 |
|
|
|
| 37 |
A |
797 |
785 |
20.43 |
|
|
|
| 38 |
A |
749 |
739 |
2.84 |
|
|
|
| 39 |
A |
605 |
597 |
17.72 |
|
|
|
| 40 |
A |
438 |
432 |
5.16 |
|
|
|
| 41 |
A |
372 |
367 |
2.29 |
|
|
|
| 42 |
A |
328 |
324 |
23.21 |
|
|
|
| 43 |
A |
263 |
259 |
36.36 |
|
|
|
| 44 |
A |
262 |
258 |
17.51 |
|
|
|
| 45 |
A |
226 |
223 |
13.59 |
|
|
|
| 46 |
A |
208 |
205 |
4.31 |
|
|
|
| 47 |
A |
137 |
135 |
6.01 |
|
|
|
| 48 |
A |
74 |
73 |
0.94 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 35920.6 cm
-1
Scaled (by 0.986) Zero Point Vibrational Energy (zpe) 35417.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
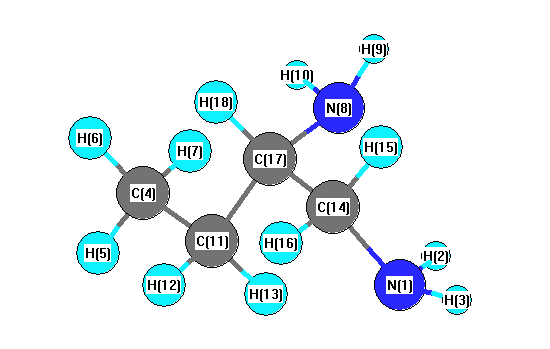
Charges from optimized geometry at B97D3/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.293 |
|
|
|
| 2 |
H |
0.122 |
|
|
|
| 3 |
H |
0.109 |
|
|
|
| 4 |
C |
-0.281 |
|
|
|
| 5 |
H |
0.086 |
|
|
|
| 6 |
H |
0.074 |
|
|
|
| 7 |
H |
0.075 |
|
|
|
| 8 |
N |
-0.305 |
|
|
|
| 9 |
H |
0.115 |
|
|
|
| 10 |
H |
0.115 |
|
|
|
| 11 |
C |
-0.086 |
|
|
|
| 12 |
H |
0.060 |
|
|
|
| 13 |
H |
0.073 |
|
|
|
| 14 |
C |
-0.060 |
|
|
|
| 15 |
H |
0.043 |
|
|
|
| 16 |
H |
0.062 |
|
|
|
| 17 |
C |
0.038 |
|
|
|
| 18 |
H |
0.053 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.086 |
1.246 |
1.168 |
1.710 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-36.499 |
0.385 |
0.799 |
| y |
0.385 |
-39.062 |
-0.368 |
| z |
0.799 |
-0.368 |
-41.249 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.656 |
0.385 |
0.799 |
| y |
0.385 |
-0.187 |
-0.368 |
| z |
0.799 |
-0.368 |
-3.469 |
|
| Polar |
|---|
| 3z2-r2 | -6.938 |
|---|
| x2-y2 | 2.563 |
|---|
| xy | 0.385 |
|---|
| xz | 0.799 |
|---|
| yz | -0.368 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.477 |
0.025 |
0.095 |
| y |
0.025 |
9.738 |
0.021 |
| z |
0.095 |
0.021 |
9.332 |
<r2> (average value of r
2) Å
2
| <r2> |
207.074 |
| (<r2>)1/2 |
14.390 |