Jump to
S1C2
Energy calculated at B97D3/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -135.144295 |
| Energy at 298.15K | -135.152384 |
| HF Energy | -135.144295 |
| Nuclear repulsion energy | 82.522460 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B97D3/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3403 |
3355 |
4.03 |
|
|
|
| 2 |
A' |
3030 |
2988 |
57.52 |
|
|
|
| 3 |
A' |
2973 |
2931 |
40.42 |
|
|
|
| 4 |
A' |
2961 |
2920 |
30.35 |
|
|
|
| 5 |
A' |
1635 |
1612 |
17.17 |
|
|
|
| 6 |
A' |
1475 |
1455 |
1.70 |
|
|
|
| 7 |
A' |
1457 |
1437 |
0.08 |
|
|
|
| 8 |
A' |
1376 |
1357 |
4.87 |
|
|
|
| 9 |
A' |
1350 |
1331 |
13.84 |
|
|
|
| 10 |
A' |
1126 |
1110 |
9.60 |
|
|
|
| 11 |
A' |
1027 |
1012 |
21.86 |
|
|
|
| 12 |
A' |
885 |
872 |
84.60 |
|
|
|
| 13 |
A' |
835 |
824 |
87.85 |
|
|
|
| 14 |
A' |
396 |
391 |
5.73 |
|
|
|
| 15 |
A" |
3483 |
3434 |
0.59 |
|
|
|
| 16 |
A" |
3037 |
2995 |
68.38 |
|
|
|
| 17 |
A" |
3001 |
2958 |
7.59 |
|
|
|
| 18 |
A" |
1466 |
1445 |
6.76 |
|
|
|
| 19 |
A" |
1366 |
1347 |
0.01 |
|
|
|
| 20 |
A" |
1248 |
1231 |
0.11 |
|
|
|
| 21 |
A" |
992 |
978 |
0.82 |
|
|
|
| 22 |
A" |
767 |
756 |
1.08 |
|
|
|
| 23 |
A" |
281 |
277 |
26.49 |
|
|
|
| 24 |
A" |
241 |
238 |
14.95 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 19905.0 cm
-1
Scaled (by 0.986) Zero Point Vibrational Energy (zpe) 19626.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B97D3/cc-pVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
-1.313 |
-0.085 |
0.000 |
| C2 |
0.000 |
0.576 |
0.000 |
| C3 |
1.217 |
-0.356 |
0.000 |
| H4 |
2.158 |
0.208 |
0.000 |
| H5 |
1.212 |
-1.003 |
0.886 |
| H6 |
1.212 |
-1.003 |
-0.886 |
| H7 |
0.039 |
1.234 |
-0.877 |
| H8 |
0.039 |
1.234 |
0.877 |
| H9 |
-1.384 |
-0.697 |
0.811 |
| H10 |
-1.384 |
-0.697 |
-0.811 |
Atom - Atom Distances (Å)
| |
N1 |
C2 |
C3 |
H4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| N1 | | 1.4701 | 2.5447 | 3.4833 | 2.8289 | 2.8289 | 2.0826 | 2.0826 | 1.0180 | 1.0180 |
C2 | 1.4701 | | 1.5329 | 2.1888 | 2.1783 | 2.1783 | 1.0970 | 1.0970 | 2.0471 | 2.0471 | C3 | 2.5447 | 1.5329 | | 1.0971 | 1.0966 | 1.0966 | 2.1643 | 2.1643 | 2.7450 | 2.7450 | H4 | 3.4833 | 2.1888 | 1.0971 | | 1.7736 | 1.7736 | 2.5117 | 2.5117 | 3.7438 | 3.7438 | H5 | 2.8289 | 2.1783 | 1.0966 | 1.7736 | | 1.7716 | 3.0796 | 2.5252 | 2.6142 | 3.1155 | H6 | 2.8289 | 2.1783 | 1.0966 | 1.7736 | 1.7716 | | 2.5252 | 3.0796 | 3.1155 | 2.6142 | H7 | 2.0826 | 1.0970 | 2.1643 | 2.5117 | 3.0796 | 2.5252 | | 1.7537 | 2.9324 | 2.3991 | H8 | 2.0826 | 1.0970 | 2.1643 | 2.5117 | 2.5252 | 3.0796 | 1.7537 | | 2.3991 | 2.9324 | H9 | 1.0180 | 2.0471 | 2.7450 | 3.7438 | 2.6142 | 3.1155 | 2.9324 | 2.3991 | | 1.6213 | H10 | 1.0180 | 2.0471 | 2.7450 | 3.7438 | 3.1155 | 2.6142 | 2.3991 | 2.9324 | 1.6213 | |
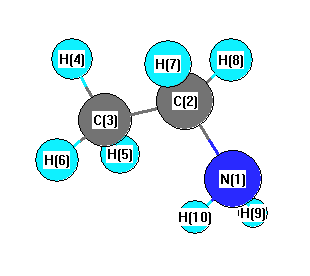 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C2 |
C3 |
115.840 |
|
N1 |
C2 |
H7 |
107.553 |
| N1 |
C2 |
H8 |
107.553 |
|
C2 |
N1 |
H9 |
109.372 |
| C2 |
N1 |
H10 |
109.372 |
|
C2 |
C3 |
H4 |
111.589 |
| C2 |
C3 |
H5 |
110.775 |
|
C2 |
C3 |
H6 |
110.775 |
| C3 |
C2 |
H7 |
109.650 |
|
C3 |
C2 |
H8 |
109.650 |
| H4 |
C3 |
H5 |
107.899 |
|
H4 |
C3 |
H6 |
107.899 |
| H5 |
C3 |
H6 |
107.754 |
|
H7 |
C2 |
H8 |
106.137 |
| H9 |
N1 |
H10 |
105.550 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.292 |
|
|
|
| 2 |
C |
-0.059 |
|
|
|
| 3 |
C |
-0.253 |
|
|
|
| 4 |
H |
0.071 |
|
|
|
| 5 |
H |
0.074 |
|
|
|
| 6 |
H |
0.074 |
|
|
|
| 7 |
H |
0.077 |
|
|
|
| 8 |
H |
0.077 |
|
|
|
| 9 |
H |
0.116 |
|
|
|
| 10 |
H |
0.116 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.833 |
-0.869 |
0.000 |
1.204 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-23.970 |
2.449 |
0.000 |
| y |
2.449 |
-20.495 |
0.000 |
| z |
0.000 |
0.000 |
-19.277 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-4.085 |
2.449 |
0.000 |
| y |
2.449 |
1.129 |
0.000 |
| z |
0.000 |
0.000 |
2.956 |
|
| Polar |
|---|
| 3z2-r2 | 5.912 |
|---|
| x2-y2 | -3.475 |
|---|
| xy | 2.449 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.944 |
0.083 |
0.000 |
| y |
0.083 |
5.256 |
0.000 |
| z |
0.000 |
0.000 |
5.021 |
<r2> (average value of r
2) Å
2
| <r2> |
58.945 |
| (<r2>)1/2 |
7.678 |
Jump to
S1C1
Energy calculated at B97D3/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -135.143699 |
| Energy at 298.15K | |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B97D3/cc-pVTZ
Geometric Data calculated at B97D3/cc-pVTZ
Point Group is Cs
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability